Improving and Accelerating Therapeutic Development for Nervous System Disorders: Workshop Summary (2014)
Chapter: 2 Drug Development Challenges
Key Points
• Drug development is a lengthy, complex, and costly process, entrenched with a high degree of uncertainty that a drug will actually succeed.
• The unknown pathophysiology for many nervous system disorders makes target identification challenging.
• Animal models often cannot recapitulate an entire disorder or disease.
• Challenges related to heterogeneity of the patient population might be alleviated with increased clinical phenotyping and endotyping.
• Greater emphasis on human data might lead to improved target identification and validation.
• There is a lack of validated diagnostic and therapeutic biomarkers to objectively detect and measure biological states.
• Unfamiliarity with current regulatory processes for investigational new drug (IND) applications can be resolved through pre-IND meetings.
NOTE: The items in this list were addressed by individual speakers and participants and were identified and summarized for this report by the rapporteurs, not the workshop participants. This list is not meant to reflect a consensus among workshop participants.
David Michelson, vice president of clinical neuroscience and ophthalmology at Merck Research Laboratories, opened the workshop by underscoring drug discovery challenges for nervous system disorders. The years- to decades-long process can be complex, and there is nearly
always a moment of uncertainty that a drug will succeed to the next phase of development. This long development pipeline faces increasing costs and additional challenges, including the lack of predictive validity of current animal models, insufficient knowledge regarding underlying mechanisms of disease, patient heterogeneity, lack of targets and biomarkers, a high rate of failed clinical trials, and regulatory challenges. To better understand how these challenges create bottlenecks in the development pipeline, William Potter, senior advisor in the Office of the Director of the National Institute of Mental Health, began with an overview of current practices.
DRUG DISCOVERY AND DEVELOPMENT PATHWAY
Potter described the process of drug discovery and development beginning with target identification and validation (see Figure 2-1). A target can be a protein, DNA, or RNA that causes or contributes to disease. Its validation consists of demonstrating that modulating the target has a therapeutic effect. Assay development follows target validation and is an objective method for screening putative compounds to determine interaction and/or modification of the target. After an assay is established, the next step is to find compounds that actively engage the target. From a pool of potential compounds, a few select leads that demonstrate a relationship between chemical structure and target-based activity in a biochemical or cell-based assay are generated.
The process of moving from target identification to lead generation is often done entirely without animal studies, said Potter. Potential compounds, for example, can be generated through binding/functional, biochemical, and cellular or cytotoxicity assays. High-throughput screening through a large compound library can identify multiple compounds. Progressing to a lead compound(s) can involve complex cellular assays, toxicological surrogate assays, biopharmacological surrogates, and surrogates for absorption, distribution, metabolism, and excretion (ADME).
Potter noted that animal models are often used first to narrow the number of lead compounds to one or two candidates that can proceed into clinical trials. The lead compound(s) is tested in animals for its pharmacological and toxicological properties. Animal tests for efficacy—as opposed to safety—are, in most cases, not required prior to first-in-human testing,
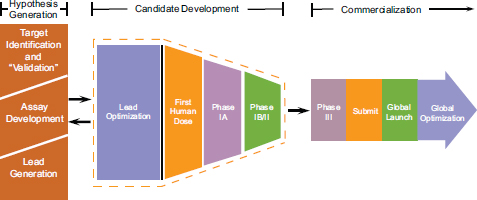
FIGURE 2-1 Overall drug discovery and development process.
SOURCE: Potter presentation, April 8, 2013.
a point repeatedly stressed by several workshop participants. After a lead compound is generated, it undergoes further testing to optimize physicochemical and pharmacological properties, especially potency and selectivity. Optimization is an elaborate process that can be costly and time-intensive. Despite the resources (e.g., time, personnel, and finances) devoted to generating lead compounds, Potter observed that many fail during optimization.
Once optimization is complete, first-in-human testing can begin with a Phase Ia clinical trial in which a single dose of the drug is given to healthy volunteers. This is followed by Phase Ib trials, which consist of multiple escalating doses to establish safety, steady-state pharmacokinetics, and maximum tolerated dose. There is increasing use of Phase Ib trials to provide evidence of efficacy in order to establish proof of concept (POC).1 Potter noted that a typical POC clinical trial is a small controlled study conducted at fewer than 4 sites with less than 100 subjects/patients. If the drug succeeds at POC, clinical trials then proceed to larger Phase II and Phase III trials, which consist of randomized, usually placebo-controlled arms, to ensure safety and efficacy (see Table 2-1).
______________
1POC refers to the minimum number of experiments/studies that provide critical data—efficacy, receptor occupancy, safety, and tolerability—to support either going forward to larger clinical trials or “killing” a compound, said Potter.
TABLE 2-1 Stages of Drug Development
| Stage | Method | Purpose |
| Preclinical | Animal, in vitro, and laboratory studies | Testing toxicity, efficacy, pharmacokinetics, and pharmacodynamics |
| Investigational New Drug Application | ||
| Phase I | Healthy human volunteers (~20–100) | Testing the safety of a single dose (Phase Ia) and multiple doses (Phase Ib) of a drug; also includes pharmacokinetics and maximum tolerated dose |
| Phase II | Patients (~100–300) | Assessing safety and efficacy |
| Phase III | Patients (hundreds to thousands; typically 1,000–2,000) | Assessing safety and efficacy |
| New Drug Application | ||
| Phase IV | Varies | Postmarketing surveillance |
SOURCE: Potter presentation, April 8, 2013.
After successful completion of Phase III and submission of a new drug application (NDA) to the U.S. Food and Drug Administration (FDA), a drug becomes eligible for marketing. Even with marketing approval, a drug continues to be studied through postmarketing surveillance to ensure safety.
CURRENT DRUG DEVELOPMENT CHALLENGES
Christopher Austin, director of the National Center for Advancing Translational Science (NCATS), highlighted underlying challenges behind translational failures that need to be addressed to improve the drug development pipeline for nervous system disorders (Wegener and Rujescu, 2013) (see Box 2-1).
BOX 2-1
Drug Development Challenges for Nervous System Disorders
• Insufficient knowledge of underlying biological mechanisms of diseases
• Translational failures using animal models
• Lack of detailed clinical phenotyping and endotyping due to high heterogeneity of patient populations
• Lack of valid biomarkers and surrogate endpoints
• Inability to replicate preclinical studies
• Inadequate collaboration among academic researchers, industry, and government
• Innovation gaps leading to an inability to address the unmet need
NOTE: The items in this list were addressed by individual speakers and participants and were identified and summarized for this report by the rapporteurs, not the workshop participants. This list is not meant to reflect a consensus among workshop participants.
Unknown Biological Mechanisms and Biomarkers of Diseases
One prominent challenge is that mechanisms behind nervous system disorders are poorly understood. Austin stated that if mechanisms of nervous system disorders were better understood, then more effective interventions could be developed; however, clinical observations made during drug development and conventional neuropharmacology should not be omitted as mechanisms for understanding these disorders. Several participants also noted that having a better understanding of the specific neural pathways involved in disease etiology may help researchers develop more comprehensive assays using emerging tools and technologies. This, in turn, may lead to the development of more targeted therapeutics. In addition, identifying biomarkers is essential, not only to provide proof of mechanism but also to refine targets, provide POC, and evaluate whether an early intervention focused on a single target can prevent or forestall disease (IOM, 2011, p. 34). According to Fleming and Powers, biomarkers are indirect measures of clinically meaningful endpoints, which are direct measurements of how patients feel, function, and survive (Temple, 1995). Although they may provide valuable information regarding effects on biological activity during an intervention, biomarkers can be unreliable predictors of clinical efficacy. Extensive
clinical trials in patient populations are needed to validate biomarkers as true surrogate endpoints, outcome measures that are used as a substitute for a clinically meaningful endpoint (Fleming and Powers, 2012).
According to Reisa Sperling, director of the Center for Alzheimer’s Research and Treatment and professor of neurology at Harvard Medical School, and Paul Aisen, director of the Alzheimer’s Disease Cooperative Study and professor in the department of neurosciences at the University of California, San Diego (UCSD), Alzheimer’s disease (AD) is a prominent example of how a greater understanding of mechanism and improved biomarkers might accelerate drug development. Both speakers commented that AD begins years, perhaps decades, prior to the onset of symptoms, but the absence of objective biomarkers continues to be a challenge. The best opportunity to intervene may be prior to symptoms, but earlier interventions decrease the likelihood of detecting clinical signals of the disease (Rowe et al., 2010). Aisen commented that the central hypothesis for AD, specifically, the amyloid hypothesis, might not be valid. Although compelling genetic data exist to support the role of β-amyloid and converging data indicate that the presence of β-amyloid increases risk of cognitive decline in prodromal and preclinical AD, there are still many unknowns, Sperling said. One unknown is whether β-amyloid itself is sufficient to cause AD, because there might be other factors necessary to initiate the disease process. Another unknown is what species of β-amyloid should be targeted, such as β-amyloid42 or oligomeric β-amyloid, or other cleavage products of amyloid precursor protein (APP). Weighing against the β-amyloid hypothesis are disappointing results from multiple anti-β-amyloid clinical trials in mild to moderate dementia, said Sperling.
If the underlying biological mechanisms of a disease are unknown, how can researchers identify targets and eventually develop drugs? How do you develop biomarkers for diseases and disorders that are difficult to define? Starting at the molecular level is important, said Austin; however, it is not a stepwise process (A to B to C) to clinical trials.
Translational Failures Using Animal Models
Many participants discussed the inability of animal models to accurately predict efficacy as a challenge to drug development. Although animal models work reasonably well to prioritize reagents for a clinically validated target, they are not as useful to prioritize reagents aimed at
novel targets, opined Chas Bountra, head of the Structural Genomics Consortium and professor of translational medicine at the University of Oxford. In addition, animal models can be poor predictors of clinical efficacy and therapeutic index. This is most likely due to animal models’ inability to fully mimic diseases, as demonstrated by a groundbreaking study of the failure of mouse models in human inflammatory diseases (Seok et al., 2013). Potential mismatch of preclinical and clinical endpoints could be another reason for translational failures, although corresponding preclinical and clinical endpoints may not be sufficient enough to predict clinical efficacy (IOM, 2013). Austin noted that there are many examples of a lack of drug efficacy in clinical trials after successful animal studies (i.e., failure of efficacy) as well as the presence of human toxicity not previously shown in animal studies (i.e., failure of toxicity). Some of these failures relate back to the lack of understanding of mechanisms for disease; how can successful animal models be created based on unknown mechanisms?
Two speakers noted that the limitations of existing animal models have resulted in translational failure. Lawrence Goldstein, director of the UCSD Stem Cell Program and distinguished professor in the department of neurosciences at the UCSD School of Medicine, highlighted several challenges related to existing animal models of AD, including the inability to develop all the symptoms of AD; overexpression of proteins linked to disease (e.g., APP) at levels high enough to produce abnormal phenotypes; transgenic mouse models that fail to fully recapitulate AD pathology (Duff and Suleman, 2004); lack of sporadic AD models, which account for 95 percent of cases (Young and Goldstein, 2012); and inability of drugs found efficacious in animal models to translate to clinical trials. Wayne Drevets, scientific vice president and disease area leader in mood disorders at Janssen Pharmaceutical Companies of Johnson & Johnson, echoed similar comments for depression (Banasr et al., 2011; Manji et al., 2001; Savitz et al., 2013), one of which is the lack of animal models for spontaneously recurring mood disorders (e.g., bipolar disorder). To summarize, Bountra noted that animal models do not always accurately predict dose, tolerability, efficacy, and research priority.
Lack of Clinical Phenotyping and Patient Stratification
Austin stated that the field has lost track of detailed clinical phenotyping, which has advanced other fields. It is important that the
field improve phenotyping of nervous system disorders, he noted. Failed clinical trials are almost predictable simply due to patient heterogeneity. From an investment standpoint, stated Kiran Reddy, principal at Third Rock Ventures, the heterogeneity of patient populations necessitates larger, more complex, and thus more expensive clinical trials. Patient populations with depression and bipolar disorder, for example, are highly heterogeneous, and segregating patients based on phenotypes may serve as a useful method of patient stratification when conducting clinical trials. Without new ways to stratify patients, physicians cannot always predict dose responses or the speed of response, noted Bountra. When planning clinical trials, it will be important to consider patient groups in which the mechanism of disease is most likely to be homogeneous. Patient stratification could produce better clinical trial outcomes and additional information about potential therapeutic targets. Austin also pointed out that a simple relationship between genotype and phenotype mechanisms does not exist. Investigators often mistakenly think that after the genes or polymorphisms are identified, the clinical effect will be understood. This is often not the case, and more research is needed to identify the relationship between phenotype and genotype mechanisms, Austin stated.
Inability to Rely on Published Data
Reproducibility remains a critical issue for translation. The ability to rely on published data and process those data from one lab to another is critical for the successful translation of discovery research, noted speakers Austin and Hodes. Investigators have finally reached the point at which success cannot be achieved independently; Austin made an analogy to the field having reached the first step in any 12-step rehabilitation program: acknowledging that an issue exists. A participant pointed out two systemic lapses that may need to be addressed regarding the current 95 percent confidence interval: (1) only top-tier journals have statistical editors to assess whether or not a p value is credible; and (2) funding agencies also require a p value of 0.05 or less for potential grantees; however, there is no assessment for statistical credibility. The field would likely benefit from publicly available data on everything from compound activity to patient registries to natural history, said Austin. In addition, explicit reporting of experimental results could increase reproducibility. Consistent with discussions from the 2012 Institute of Medi-
cine animal models workshop, the scientific validity and reproducibility of preclinical data are important when making decisions regarding when to go into clinical trials (IOM, 2013).
Inadequate Collaboration Among Academia, Industry, and Government
Bountra said that organizational challenges are formidable in the drug development process. The greatest challenge is that the majority of the field is working competitively on the same few molecular targets. Bountra noted that many academic laboratories are interested in researching disease networks and biological pathways, but have limited access to reagents, such as inhibitors and antibodies. Because most drug testing fails in Phase IIa and because the negative results are usually not shared, the field is potentially wasting resources (e.g., time and money), said Bountra. As mentioned previously, there is a shift in focus of research and development groups; industry is successful at processes that require scale and infrastructure (e.g., high-throughput screening, lead optimization, and manufacturing), noted Bountra, whereas academia continues to be a source of new knowledge in biomedical research needed for early drug discovery (Bunnage, 2011).
Pipeline Challenges
Reddy expressed the view that there are several pipeline problems plaguing large pharmaceutical companies. During the past 15 years, companies have steadily increased expenditures on research, but the number of new drug approvals has dipped (see Figure 2-2). Adrian Ivinson, director of the Harvard NeuroDiscovery Center at Harvard University, noted that during this timeframe, only a small handful of nervous system drugs were approved, despite a growing market coupled with unmet need.
At a recent life sciences conference, venture capitalists were surveyed about which therapeutic areas in the life sciences offered the most promising investment opportunities, said Reddy. Oncology and immunology were seen as the best areas of opportunity by 30 and 17 percent of respondents, respectively. Nervous system disorders fell significantly behind, with only 8 percent of investors viewing the field favorably.
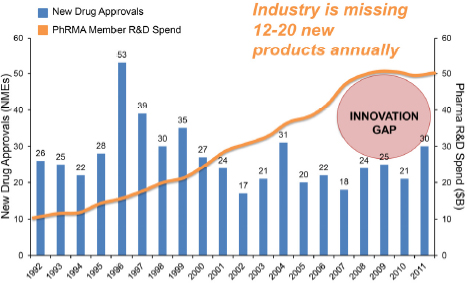
FIGURE 2-2 Opportunity for venture capital firms to fill the innovation gap.
NOTE: NME = new molecular entity; PhRMA = Pharmaceutical Research and Manufacturers of America; R&D = research and development.
SOURCE: Reddy presentation, April 9, 2013.
Kazumi Shiosaki, managing director at MPM Capital, noted that when her organization looks to invest in a particular drug, it uses a checklist of questions to gauge the risk of investment, including
• Has the drug target been identified (versus a drug identified in a phenotypic screen)?
• Has the target been validated as a way to arrest the disease?
• Are the biochemical interactions of the drug candidate known?
• Is there information about dose dependence in animal models?
• Has safety of administration on a chronic basis been shown?
• Can the drug cross the blood–brain barrier?
• Do toxicology studies show it is a safe drug?
• Is there a sufficient therapeutic window?
• Are drug purity and stability acceptable?
• Is there good protection of intellectual property?
Multiple challenges can impact the drug development pipeline, originating with the lack of understanding of underlying biological mechanisms of nervous system disorders. Lawrence Goldstein suggested that the field identify key bottlenecks in the pathway and become better at
tolerating a certain amount of uncertainty and risk to improve therapeutic development.
THE REGULATORY PROCESS
Several speakers from FDA relayed their individual perspectives on the regulatory process. Speakers emphasized that IND applications are reviewed on a case-by-case basis and that researchers can use pre-IND meetings as a resource to answer technical questions prior to submission. To begin the conversation, Imran Khan, pharmacologist and toxicologist in the Office of New Drugs, Center for Drug Evaluation and Research of FDA, provided an overview of the review process for IND applications; common inadequacies that could put applications on clinical hold, an FDA-ordered delay or suspension of clinical trials3; and how issues can be mitigated.
To initiate clinical trials, IND regulations4 state that the drug’s sponsor first must submit adequate evidence from pharmacological and toxicological studies in laboratory animals or in vitro studies that supports the conclusion that the proposed clinical trial is reasonably safe. The application would need to contain evidence of the drug’s chemistry, manufacturing, and controls (CMC) (details of product manufacturing, product stability, and shelf-life), and preclinical studies of pharmacology, pharmacokinetics (PK), and ADME. Results from acute, sub-chronic, and chronic toxicity tests, including reproductive and developmental toxicity and special toxicity testing related to the drug’s mode of administration or conditions of use, would be included. The data requirements for drugs can vary according to their clinical experience or history: new molecular entities (NMEs) never tested in humans, NMEs with prior human experience, and marketed drugs for which the sponsor seeks a new indication.
In the case of an NME never tested in humans, pharmacological studies require inclusion of in vitro receptor binding screens and in vitro pharmacodynamic (functional) assays. However, Khan noted that animal efficacy models are not necessary at this stage. Under safety pharmacology, a core battery of studies is required to assess the effects of the drug on vital organs, such as those within the cardiovascular, respiratory, and central nervous systems. PK must be conducted in the same animal spe-
______________
3See http://www.fda.gov/downloads/AboutFDA/CentersOffices/CDER/ManualofPoliciesProcedures/ucm082022.pdf.
421 C.F.R. 312.22 and 21 C.F.R. 312.23.
cies used for toxicity testing. Also required are tissue distribution and in vitro metabolism data in microsomes and/or hepatocytes from humans and animals. Toxicological studies are required to be conducted in two species, a rodent and a non-rodent—usually dog, monkey, or guinea pig—of similar or longer duration than the proposed human exposure. The genotoxic potential5 of the drug is required to be studied in a battery of in vitro and in vivo assays for determining mutations and chromosomal damage. Impurities with genotoxic potential may also need to be assessed for genetic toxicity.
In the case of an NME with previous human experience, Khan stated that some of the studies outlined above may not be required. For example, new safety pharmacological data are not needed if adequately documented safety data with previous human experience are included. However, the intended duration of drug administration must be supported with comparable or longer repeat-dose toxicity studies. Furthermore, genotoxicity, fertility, and reproductive studies may also be required. For example, studies of fertility in early embryonic development and ADME in fetal development must be completed prior to Phase III, and pre- and post-natal development studies completed prior to submission of the NDA.
In the case of an already-approved drug for which a new indication is sought, most of the non-clinical studies are not required, said Khan, as long as the following are true: the proposed dose and duration are consistent with those of the approved product; the intended route of administration must be the same as the marketed product; the patient population must be similar to that for which the product has been already approved; and the predicted PK/ADME are sufficiently similar to the marketed product.
Khan noted that biologics are regulated differently from small molecules in the following ways:
• Biologics may not need to be tested for genotoxicity.
• Biologics may not need to be tested in two species, because sometimes only one species is pharmacologically relevant.
• Biologics may not need to be tested for antidrug antibodies, especially if no toxicity is observed at an adequately high dose.
• The criteria for an adequately high dose may be different.
______________
5The drug’s ability to cause genetic damage.
• The assessment of pharmacodynamic effects in toxicity studies may be helpful.
• Acute-dose toxicity studies may not be adequate to support a single-dose clinical trial if a long half-life of elimination in humans is anticipated.
Clinical Holds
Khan noted that many common reasons can lead to a clinical hold of an IND application. Non-clinical studies that are irrelevant or inadequate are two possible reasons. Inadequate studies might lack sufficient documentation, test too few animals, not assess standard parameters or not provide data, study inadequate doses, use a route of administration other than that proposed for the humans without justification, and have had an insufficient duration. Another common reason for a clinical hold is that a no-effect dose for serious toxicity was or could be determined. Serious toxicity with no or an inadequate strategy for monitoring the toxicity in humans could warrant a clinical hold as well. In addition, problems with CMC that require non-clinical safety testing (e.g., impurities associated with genotoxicity) and a novel excipient6 in the clinical formulation that has not been adequately tested in animals are problematic. Lastly, if the clinical investigator has an inadequate brochure, then that could lead to a clinical hold, said Khan.
In some cases, a clinical hold may be avoided, said Khan. For example, a sponsor can revise the clinical protocol to limit the dose to provide a sufficient safety margin (e.g., one-tenth of the plasma Cmax at the no observed adverse effect level [NOAEL] for convulsions in animals). The sponsor can also limit the duration of dosing, ascend doses slowly with careful monitoring, or limit the drug’s duration if the toxicity occurs after prolonged administration. In other cases, the sponsor can submit scientific justification to support the original clinical protocol. For example, for inadequate high-dose selection in a pivotal toxicity study, the sponsor can provide data to document that the toxicity is not relevant to humans. Khan emphasized that when protocols depart from standard testing requirements, justification is necessary.
______________
6An inactive ingredient that is formulated with a therapeutic (e.g., solvents, fillers, and flavors) (FDA, 2005b).
Guidance and Clarification for IND Applications
Following Khan’s overview, Eric Bastings, deputy director of the Division of Neurology Products in the Center for Drug Evaluation and Research (CDER) at FDA, and Ni Khin, medical team leader in the same division, joined Khan to field questions from participants related to the regulatory process. When asked to explain the animal rule for IND applications, Bastings explained that animal studies are needed only when human efficacy trials are neither feasible nor ethical. In some cases, a risk–benefit analysis is needed for protocols in which animal efficacy models may be needed. Bastings added that, typically, animal models are not required for INDs to be tested in clinical trials. Bastings and Khin stressed the importance of case-by-case decision making regarding regulatory requirements and reiterated the importance of a pre-IND meeting for discussion of the elements of a successful IND.
Several participants inquired about the regulatory process for therapeutics that could potentially treat serious or life-threatening disorders. Bastings noted two programs designed to expedite FDA’s review process in these cases. The first is known as Fast Track, which applies to drugs intended for serious diseases or that fill an unmet need. The unmet need criterion is defined as providing therapy where none exists or providing therapy that may be potentially superior to existing therapies (FDA, 2013b). The process for receiving a Fast Track designation is expedited in the following ways: more frequent meetings with FDA, more frequent written correspondence, and a rolling review process, which allows a sponsor to submit portions of its NDA before the rest of its submission. Normally, FDA requires that the entire package be submitted together.
The second FDA program to expedite regulatory review is through a “breakthrough therapy designation.” Breakthrough therapies must meet two criteria: be intended for serious or life-threatening conditions, and demonstrate preliminary clinical evidence that the drug may have substantial improvement over available therapy (FDA, 2013a). Breakthrough therapies can take advantage of all the features of the Fast Track designation plus more intensive guidance from FDA on an efficient drug development program. These additional features include holding meetings throughout the different drug development processes, ensuring that the design of clinical trials is as efficient as possible, and assigning a cross-disciplinary project lead for the FDA review team to facilitate review.
Many participants noted that there is complexity and a lack of clarity in the IND application process. Khin directed participants to the FDA website to find guidance on the process; however, Khin noted that the guidance is non-binding. A participant stated that there has been a transition from traditional efficacy studies using animals to select efficacy studies when there are sufficient safety data and a plausible mechanism. The field is developing tools to better understand the molecular level of a disease and move toward a patient-stratified approach with the goal of developing compounds with less toxicity. The participant concluded with an open question about whether the IND application process would be different for these cases.
Strategies to Improve Communication and Reduce Regulatory Delays
Robert Conley, regulatory leader of Biomedicines at Eli Lilly and Company, offered several thoughts on how to improve communication and reduce regulatory delays. Conley first observed that startups and small drug companies seem to be afraid of communicating with FDA. One way to reduce fear and improve understanding is to meet with FDA at pre-IND meetings. These meetings can help to explain the basis for regulatory requirements.
Next, Conley observed that increased harmonization of requirements among FDA divisions might lead to a better understanding of requirements by applicants and, therefore, higher rates of and faster times to acceptance. Problems with conflicting or divergent requirements occur most frequently with drugs for which a new indication is sought. For example, the safety pharmacology requirements might differ between nervous system drugs and cardiovascular drugs, making it necessary for the sponsor to conduct an unexpected new round of pharmacological testing. The time and expense might be too great for a fledgling sponsor. If an impasse is reached between FDA and a sponsor, then it is important to learn from the experience, said Conley.
Conley’s third observation was to point to the cultural disconnect between FDA and the National Institutes of Health as a potential reason for delays in the approval process. Academics sometimes approach FDA as though they are the “experts” and, because of extensive peer review, believe less oversight is needed. Laws, regulations, and guidance drive
FDA culture; it is not driven by the standards of merit that typically apply to the academic community.
Finally, Conley suggested that academic and industry colleagues come to any FDA meeting with a “Plan B” in order to encourage resolution of differences and decrease delays. Conley asserted that academics and industry tend to argue too vehemently for their own points without understanding the constraints under which FDA operates.
The current drug development pipeline is multifaceted and complex, composed of great risk that a drug will not be successful even after many resources have been invested. Nervous system disorder drugs in particular face a number of challenges that complicate drug development even further. Although regulatory processes are not intended to hinder drug development, many investigators are unclear of the specific requirements for INDs and request comprehensive guidance. Opportunities to improve and accelerate drug development for nervous system disorders through emerging new tools and technologies, novel methodological approaches, and infrastructural changes is explored in subsequent chapters.















