Sickle Cell Disease in Social Security Disability Evaluations: Pain and Treatment Settings (2025)
Chapter: 2 Pain in Sickle Cell Disease
2
Pain in Sickle Cell Disease
Pain is a hallmark of sickle cell disease (SCD), contributing to morbidity, reduced quality of life, and disability of individuals living with the disease (Ballas and Darbari, 2020; Platt et al., 1991; Shah et al., 2019). By definition, pain from any condition is a subjective experience (Wideman et al., 2019), and there is tremendous heterogeneity in pain symptoms, including their quality, duration, and locations; timing; and associated co-morbid conditions among those with SCD (Dampier et al., 2017; Field et al., 2019; Glaros and Brandow, 2020). In this chapter, the committee discusses the etiology, diagnosis, experiences, and timing of acute and chronic pain in people with SCD. Chapter 3 will discuss treatments for both acute and chronic pain.
TERMINOLOGY
Before the molecular and genetic origins of SCD were known, people and families in Africa referred to the disease based on the debilitating pain it caused. For example, in Ghana, SCD was known as chwech-weechwe or ahotutuo, meaning “relentless, repetitive gnawing pain” or “pain in bones and joints.” Today, people living with SCD, their families, health care providers, and researchers commonly refer to painful episodes as “crises,” while the SCD literature often uses the phrase “vaso-occlusive crises.” International Classification of Diseases (10th version; ICD-10) diagnostic codes exist for “vaso-occlusive crisis” or “SCD with crisis.”
ICD-10 codes provide the option to specify other types of vaso-occlusive complications, such as acute chest syndrome, splenic sequestration, hepatic sequestration, priapism, and dactylitis. These complications can be painful, but clinicians typically consider them as distinct complications of SCD, or “complicated crisis.”
Given this lack of clarity, there is movement away from using the phrase “sickle cell crisis.” There is also an effort to acknowledge and treat pain without catastrophizing it (Schneider et al., 2022), as increased negative thinking about pain by individuals’ self-report is associated with increased pain scores and higher opioid requirements (Citero et al., 2007; Finan et al., 2018). Some practitioners have concerns that seeking care for a “crisis” might suggest to uninformed practitioners that persons with SCD are not actively managing their disease and are presenting “in crisis.” As such, many advocate for the more neutral phrase “vaso-occlusive episode.” Others prefer to exclude the qualifier of vaso-occlusion too, because of the emerging concept that not all pain exacerbations result from incurrent vaso-occlusion and nociceptive pain. Therefore, many clinicians, researchers, and individuals with SCD may use the terms “painful episode” or “sickle cell-related pain.”
In keeping with the published literature and diagnostic codes used in the medical records available to the Social Security Administration, the committee has chosen to use the phrases “pain crisis” to refer to acute or acute-on-chronic episodes of pain above baseline and “chronic pain” to refer to persistent pain that is experienced daily or on most days by some individuals with SCD. Given that pain, in general, is an underlying source of impaired function and disability, regardless of whether it is acute or chronic, the committee may also frequently just refer to “pain.”
ACUTE PAIN PATHOPHYSIOLOGY
The etiology of pain in SCD is complex and multifactorial (Ballas and Darbari, 2020; Field et al., 2019; Kavanagh et al., 2022; Takaoka et al., 2021). As the phrase vaso-occlusive crisis suggests, an inciting event is blood vessel occlusion caused by sickle-shaped red blood cells becoming lodged in post-capillary venules (Diggs and Ching, 1934; Manwani and Frenette, 2013; Sundd et al., 2019; Yale et al., 2000) (Figure 2-1).
Microvascular occlusion results in ischemia—a reduction in blood flow, oxygen, and nutrients to the tissues—and subsequent ischemia-reperfusion injury, both of which are extremely painful. This pain often occurs in the bones, bone marrow, and joints but may also occur in the muscles and muscle fascia, spleen, liver, lungs, and potentially the reproductive organs. Inflammation and oxidative stress follow vaso-occlusion, resulting in the release of white blood cells, cytokines, and increased expression of vascular
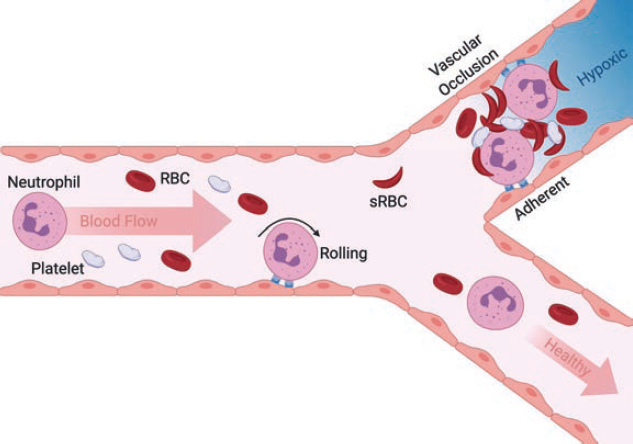
NOTE: Neutrophil rolling to arrest on inflamed endothelium is a normal innate immune response to tissue injury. Adherent neutrophils can also capture sickled red blood cells and platelets in post-capillary venules leading to vascular occlusion (upper bifurcation). Dramatic reduction in blood flow and lack of oxygen results in hypoxia and episodic pain, and often leads to organ failure. When inappropriate neutrophil activation and adhesion is absent and secondary capture of [red blood cells] RBCs and platelets limited, the vasculature remains perfused and healthy (lower pathway).
NOTE: RBC = red blood cell; sRBC = sickled red blood cell.
SOURCE: Morikis et al., 2021, Figure 1. CC BY 4.0.
adhesion molecules, creating a proinflammatory and damaging environment to local tissues and nerves that perpetuate pain even after the inciting occlusion has resolved (Jang et al., 2021).
Tissue damage in this inflammatory environment and ischemia affecting nerves can lead to peripheral neuropathy. This neuropathic component of pain is often underdiagnosed and undertreated in persons with SCD (Brandow et al., 2014; Sharma and Brandow, 2020; Sigalla et al., 2021). Occasionally, severe ischemia will lead to infarction or necrosis (i.e., cell or tissue death), particularly in areas of bone with limited vascular supply, such as the head of the femur and humerus (Ansari and Gavins, 2019; Chacko et al., 2013; Novelli and Gladwin, 2016).
The frequency and severity of these pain crises can vary widely among individuals, influenced by their unique genetic, environmental, and psychosocial factors (Field et al., 2019). The current paradigm for managing acute
pain in SCD is through analgesic medications that allow the individual to tolerate the pain, but these treatments do not reverse, correct, or affect the underlying disease process.
CHRONIC PAIN PATHOPHYSIOLOGY
Repeated acute nociceptive input from vaso-occlusion can lead to central sensitization—hyperexcitability of the central nervous system—and central nervous system inflammation, leading to long-term alterations in pain processing. This hypersensitivity lowers the threshold at which an individual perceives pain, such that pain signals from painful stimuli are amplified and non-painful stimuli are perceived to be painful. As a result, an individual can perceive pain even in the absence of evidence of tissue injury, damage, or inflammation from vaso-occlusion. This combination of recurrent acute pain episodes and central sensitization can result in a transition from acute to chronic pain in individuals with SCD. Chronic pain, defined as pain on most days of the month for six months or more (Dampier et al., 2017), is developed and maintained from central sensitization and neuronal inflammation from ongoing tissue damage (Pas et al., 2018).
Three subtypes of chronic SCD pain have been defined: (1) chronic SCD pain with contributory disease complications, such as avascular necrosis and leg ulcers; (2) chronic SCD pain without contributory disease complications, including persistent pain in the absence of evidence of tissue damage or injury; and (3) mixed pain subtype, such as widespread pain with evidence of SCD disease complications (Dampier et al., 2017). Chronic pain in SCD is often associated with disease-related complications such as bone infarction, avascular necrosis, compression fractures, osteomyelitis, and leg ulcers, but it can occur in the absence of these disease complications (Brandow et al., 2017; Niscola et al., 2009).
While it is likely that chronic SCD pain involves a combination of nociceptive, neuropathic, and nociplastic (formerly known as central) mechanisms, nociplastic pain driven by central sensitization is believed to be the predominant mechanism of pain for individuals with chronic SCD pain without contributory disease complications. Individuals with this pain subtype report widespread pain that exacerbates psychological comorbidities and have been noted to have higher opioid consumption and pain interference (Kuisell et al., 2023; Levenson et al., 2008). However, more studies are required to understand the prevalence of nociplastic pain among individuals with chronic SCD pain. Bone damage is thought to be another important factor contributing to chronic pain in SCD, though the association between chronic pain and bone damage is poorly understood despite abundant radiographic evidence of bone damage in adults with SCD.
Vaso-occlusive crises often increase in frequency and intensity with age among children and young adults, and, by late adolescence and early adulthood, individuals with SCD experience persistent pain between vaso-occlusive crises or even in the background of a vaso-occlusive crisis. Studies indicate that between half and two-thirds of adults with SCD experience chronic pain more than half the time (Jagtiani et al., 2024; Lanzkron et al., 2018; Matthie et al., 2020), and nearly 30 percent experience pain on more than 95 percent of the days surveyed (Smith et al., 2008). An estimated 20 to 40 percent of adolescents and even younger children with SCD report persistent, daily pain (Dampier et al., 2017; Sil et al., 2016a,b).
People with chronic pain can experience exacerbations of their chronic pain and identify distinctly separate acute crises, which may prompt different approaches to pain management. While teasing apart the patterns helps to inform decision making about pain management, the distinctions between acute, acute-on-chronic, and chronic pain may be irrelevant to the individuals experiencing the pain. Ultimately, it is the individual’s experience of pain, regardless of type, and its effect on their ability to function that are important.
EXPERIENCE OF PAIN
Individuals with SCD describe acute pain crisis as sudden, intense, agonizing, continuous, inescapable, limitless, and excruciating pain that can feel like a sharp stabbing sensation, throbbing ache, or burning (Coleman et al., 2016). It can occur anywhere in the body, but most commonly pain occurs in the extremities, back, chest, and abdomen. This may result in secondary muscle tightness from overuse of some muscle groups, further exacerbating pain.
“For me, the pain feels like slamming a finger in the door or experiencing a continuous charley horse. It is excruciating. I feel like I’m thrashing inside my body. It is hard to cope with mentally, and it can go on for days to weeks to months.”
—Teanika H., adult living with SCD1
“A doctor once told me when in sickle cell crisis, it is like having labor pains times 10. It is the worst.”
—Tristan L., adult living with SCD2
___________________
1 Presented at the “Pain Management and Sickle Cell Disease – public webinar” on February 7, 2025.
2 Presented at the “Hospitalization and Sickle Cell Disease – public webinar” on December 20, 2024.
In one qualitative study using interpretative phenomenological analysis, which “allows researchers to focus on the subjective meaning and how people make sense of their experiences” (Smith et al., 2009), adults with SCD described an acute pain crisis as outside the normal parameters of pain and gave detailed descriptions of the nature of SCD pain (Coleman et al., 2016). One participant described their worst pain as having “no cutoff point” and “like being set on fire continuously.” Another participant reported that “it is just like somebody is hammering inside your bone and drilling,” and a third described it as a “woodpecker that goes ddddddddddddddd in the blood stream.”
TIMING AND “PHASES” OF VASO-OCCLUSIVE CRISES
Pain crises in SCD can be variable in both timing and intensity. Traditionally, SCD crises were thought to evolve through four phases: prodromal, initial, established, and resolving. These phases were promoted among health care providers to help them “manage (pain) according to rational basis and avoid the conflicts that often arise about the authenticity of pain” and attempt to add objectivity to the subjective experience of pain (Ballas et al., 2012). Further research revealed that the evolution of a pain crisis is neither this predictable nor linear for many individuals, particularly regarding time until resolution (Ballas et al., 2012).
The prodromal phase is reported by many as a sensation or slight discomfort that precedes full on pain. The rapper Prodigy, who lived with SCD, described it in the following way:
A sickle-cell attack would creep up slowly in my ankles, legs, arms, back, stomach and chest. Sometimes my lips and tongue turned numb, and I knew I was going into a crisis. (Hawkins, 2017)
In one study of approximately 100 patients, 59 adults with SCD reported a noticeable “premonition” one or two days before the onset of a crisis (Murray and May, 1988). If their schedules and resources allow it, many individuals attempt to rest, increase fluid intake, stay warm, or try oral analgesic medications during the prodromal phase in an attempt to stave off a full crisis. If such interventions are effective, they may avoid a crisis that requires acute care or hospitalization, but this self-care may require staying home rather than attending work, school, or social activities.
The initial and established phases of pain often require increasing analgesic medications to help an individual tolerate the pain, but pain medications do not treat or reverse the underlying pathophysiologic process. Early studies defined a painful episode as lasting more than two hours (Platt et al., 1991). Other studies have reported the average hospital stay for a
painful episode is between 9 and 11 days, with pain being the most severe at day three (Ballas et al., 2012). In a study of people with SCD keeping pain diaries, the shortest reported episode of severe pain lasted only a few hours, with the longest reported episode lasting eight weeks (White et al., 2021). Because the majority of SCD pain is treated at home, the length of hospitalization does not define the length of the crisis. Moreover, because many individuals may seek to get control of the pain in the emergency department or during a short admission and continue pain mitigation at home, specifying at least 48 hours inside the hospital as a criterion of pain crisis severity is not clinically meaningful.3 The resolution of pain also varies dramatically between individuals and pain crises.
“When [the pain is] acute I get hospitalized, and that stay could be anywhere from 3 days to 3–4 weeks.”
—Adult living with SCD4
Many health care providers do not advise continued hospitalization until the pain is completely resolved because increased length of hospitalization exposes individuals to excess hospital-associated risks, such as hospital-acquired infections and venous thromboembolism. Rather, the goal prior to discharge is often adequate pain control on an oral regimen. As such, most individuals are discharged to continue self-care at home, often with oral opioids, so they are often still unable to attend work or school for many days post-discharge. Despite provider notes or other social supports, many jobs and educational programs do not accommodate these absences (Gordon et al., 2024).
Some individuals return to a baseline of no pain after a crisis, but others continue to have pain on many days between crises that is similar in quality and distribution to a crisis but may vary in intensity (Figure 2-2). The likelihood of pain between crises increases with age and is reported by 10 percent of children with SCD and 50 percent of adults with SCD (Ballas et al., 2012). Frequently, there may be a resurgence of pain after initial resolution that may require re-hospitalization. Studies have found that about 20 percent of people with SCD have new crises within one week of a prior crisis, and 50 percent have recurrence within one month (Ballas et al., 2012). Therefore, a separation of 30 days between crises is an arbitrary cutoff for a new crisis, is not scientifically justified, and may lead to an underestimation of the impact of pain on an individual with SCD and may lead to worsened care.
___________________
3 Much of the present report focuses on uncomplicated SCD crises, which primarily entail pain. Complicated crises, which include conditions such as acute chest syndrome, multisystem organ failure, strokes, and sequestration crises, involve more than pain and can have long and complicated hospitalizations.
4 Excerpt from Call for Perspectives Response.
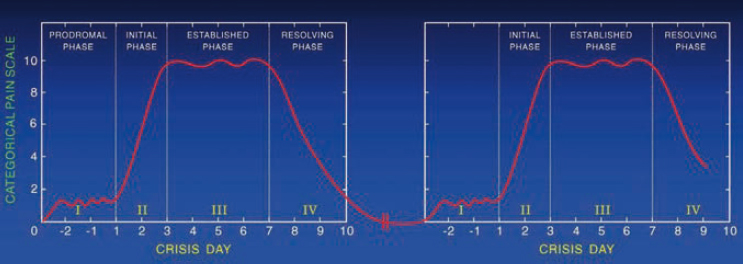
SOURCE: Ballas et al., 2012, p. 3652, Figure 4. Reprinted with permission from Elsevier.
In addition, the distinction between exacerbations of chronic pain and acute crises is difficult and seemingly arbitrary. From a patient-centered and pragmatic perspective, this is patient-defined. It is immaterial to an individual’s experience of pain, its effect on their functioning, and the underlying disease severity whether the pain is acute, acute-on-chronic, or chronic. Similarly, it is unimportant in terms of the severity of the pain or the underlying disease as to whether a pain event requiring emergency department or inpatient care occurs multiple times within a specified time period, such as 30 days; stems from a new pain crisis or complication, such as stroke; or is an exacerbation of the same pain crisis.
QUANTIFYING THE BURDEN OF PAIN
Most people with SCD will experience pain, with many experiencing repeated episodes (Platt et al., 1991). In addition, a meaningful portion of adults living with SCD experience chronic pain, as noted above. Pain is the leading cause of inpatient hospitalization for SCD, with 96 percent of admissions for SCD involving a pain crisis (Fingar et al., 2019). Early epidemiologic studies counted episodes of pain based on the number of presentations to hospital and found that pain was prevalent, but that there was an outlier group facing an increased burden, with 5 percent of people having more than three episodes per year and accounting for 33 percent of all episodes (Platt et al., 1991). The investigators conducted this study at a time when many patients did not survive to adulthood, so these findings do not reflect the currently understood burden of pain. Nonetheless, people with greater than three episodes of SCD pain per year were categorized as having severe disease as it was associated with increased mortality (Platt et al., 1991).
More recently, a prospective study comparing outcomes of people with SCD found that 54 percent of participants with HbSS disease and 46 percent
with HbSC disease had three or more acute visits to an emergency department or specialty infusion clinic over the study period (Lanzkron et al., 2018). However, the frequency of acute care utilization for pain underestimates the true burden of pain in individuals with SCD. In a prospective cohort, people with SCD over 16 years of age who kept pain diaries reported experiencing pain during more than half of all days (54.5 percent), but only reported health care utilization on 3.5 percent of days during which they reported a crisis, with many patients opting to manage their worsening pain at home (Smith et al., 2008). Studies such as this highlight that quantifying the burden of an individual’s pain should not be based solely on frequency of health care utilization.
“As a child, you want to keep up with your peers and everybody else. I would be at a 7 out of 10 pain and not even say anything. I would wait for it to get really, really bad and have to spend two weeks in the hospital.”
—Teonna W., adult living with SCD5
TRIGGERS
Pain from SCD can be unpredictable and can increase with exposure to certain triggers. Weather changes, cold temperature, dehydration, viral illness, and menstruation are all known triggers for increased pain (Borhade et al., 2024; Desai et al., 2022; Takaoka et al., 2021). A study of patient-reported outcomes from the Sickle Cell Disease Implementation Consortium found that increased pain was significantly associated with increased age, lower educational attainment, and unemployment. Because pain and decreased function have a cyclical effect on each other, it is impossible to determine from this study if increased pain is the barrier to engagement with school and work or if the stress of navigating society with these social drivers leads to increased pain (Knisely et al., 2020). Smaller studies, however, have demonstrated that anxiety, depressive symptoms, somatization, and pain catastrophizing are associated with increased sensitivity to experimental pain stimuli in people with SCD, suggesting these psychological states modulate the perception of pain in this population (Bakshi et al., 2017; Campbell et al., 2016). Furthermore, increases in perceived discrimination from health care providers, a specific source of anxiety, are associated with increased pain among adults with SCD (Haywood et al., 2014). Patients attribute this perceived discrimination to health care providers’ prejudices about the patients’ race/ethnicity and about SCD itself. Increasingly, the bidirectional interactions between SCD pain and physical or psychosocial stressors are being recognized, and there are efforts
___________________
5 Presented at the “Hospitalization and Sickle Cell Disease – public webinar” on December 20, 2024.
among health care providers to mitigate the added stress of discrimination in the health care system (Power-Hays and McGann, 2020; Reich et al., 2023). Overall, robust data exist to support the idea that the environments in which people with SCD are born, grow, live, work, are cared for, and age affect the severity of disease and overall levels of pain.
DIAGNOSIS
Despite the complex pathophysiology of pain in SCD, the nuances of which research is still elucidating, there is no diagnostic test or vital sign that clinicians can use to reliably measure the presence or severity of acute pain crises. Because SCD is a condition with no specific phenotypic traits that indicate that a person has the disease, many people with SCD describe it as an invisible disease. In addition, the potential to appear “well” while simultaneously experiencing debilitating pain can hinder treatment. People with SCD learn to distract themselves from the pain they experience (Anie and Green, 2015; Gil et al., 2001; Sil et al., 2021). Such distractions may include activities such as reading, listening to music, looking at one’s phone, or sleeping, which health care providers often perceive as “atypical” or inappropriate coping behaviors for someone experiencing debilitating pain.
There are efforts to find objective indicators of SCD pain, although research has yet to identify a clinically meaningful measure. Many studies have sought to identify laboratory markers that may indicate severity of pain or provide insight into treatment pathways (Albo et al., 2020; Douglas, 2016). Recent studies have focused on using machine learning techniques to understand the relationship between vital sign changes and pain intensity, although this is a nascent area of study (Padhee et al., 2021). While vital sign changes may be associated with a crisis, they are not sensitive or reliable markers of pain in people with SCD, and their absence does not mean the absence of pain. In addition, traditional patient-reported pain scales often have limited clinical utility, given the severity of SCD-related pain and the contextual factors that contribute to pain assessment, such as interpersonal bias (Collins et al., 2022). Thus, patient report remains the gold standard diagnostic criterion for determining an SCD crisis.
“When I’m in crisis and I hear ‘what’s your pain score,’ it doesn’t matter because I am at a thousand, but I never say that because a provider is not going to believe you.”
—Teanika H., adult living with SCD6
___________________
6 Presented at the “Pain Management and Sickle Cell Disease – public webinar” on February 7, 2025.
Ultimately, the diagnosis of an SCD pain crisis relies on the health care provider believing the patient. As such, a provider’s lack of knowledge about the disease or potential prejudices about race or opioids may limit the timely and accurate diagnosis—and hence treatment—of sickle cell pain. Unfortunately, disease-based stigma and discrimination, independent of race, and the negative perceptions held by nurses and physicians about people with SCD are widespread and well documented (Haywood et al., 2014; Jenerette et al., 2015; Reich et al., 2023). Health care providers report that negative attitudes about people of Black race affect the care of people with SCD (Nelson and Hackman, 2013). Emergency department and inpatient staff report believing incorrectly that patients with SCD are more likely to have opioid addiction and exhibit “red-flag” behaviors for drug seeking, such as requesting specific doses of specific medications, especially a specific dose of an opioid (Shapiro et al., 1997; Waldrop and Mandry, 1995). Health care providers often accuse patients with SCD of being drug seekers and treat them as though they have opioid use disorder, despite lower prevalence of opioid use disorder among those with SCD than in the general population (Liu et al., 2024; Ruta and Ballas, 2016). This is further compounded by racial disparities in pain treatment, which can result in undertreatment (Kissi et al., 2022). To mitigate delays in diagnosis and treatment, people with SCD will often prefer home pain management to avoid going to the emergency department or go through extra effort to present themselves in a way to combat these stereotypes (Phillips et al., 2023).
“In the emergency department, medical providers do not know your story or who you are. All they see is a person coming in, in a lot of pain, who probably isn’t the nicest, especially in a sickle cell crisis. They are demanding to access pain management, and medical providers think ‘oh they are here drug seeking’ or ‘they are not really in pain, not crying.’ What a lot of providers in the [emergency department] do not understand is when we are in pediatrics, we are taught how to distract ourselves, whether that is reading a book, playing a game, singing a song, [or] writing a poem. So, when you come in and you’re not screaming and making noise, but you know how much medication you need because you are trained as a child and take that over into adulthood, you get the stigma of being African American and asking for copious amounts of drugs during an opioid epidemic.”
—Teanika H., adult living with SCD7
___________________
7 Presented at the “Pain Management and Sickle Cell Disease – public webinar” on February 7, 2025.
FUNCTIONAL IMPACT
In 2020, an international team of investigators reported their findings from the multi-country Sickle Cell World Assessment Survey, which solicited input from 2,000 patients aged 12 and older and 300 health care professionals to assess overall disease burden (Osunkwo et al., 2021, 2022). Patients who completed the survey reported experiencing substantially more vaso-occlusive crises than had been reported in the literature, implying that patients under-report vaso-occlusive crises, that providers do not ask patients enough about crises, or both. About 41 percent of the patients reported that acute pain crises had a high impact on their family and social life, while 38 percent reported feeling anxious and 39 percent reported feeling depressed. More than half of the patients said their income would be higher if they did not have SCD and that SCD had a high impact on their school achievement. About half of the patients had reduced their working hours and 32 percent had been dismissed from their jobs. Survey respondents also reported they missed an average of seven hours of work because of SCD (Osunkwo et al., 2021).
Chronic pain in SCD can have a significant effect on daily functioning, health care utilization, and behavioral health, including anxiety and depression (Jonassaint et al., 2016). In children, chronic SCD-associated pain can contribute to moderate-to-severe levels of functional disability, recurrent school absences, depression, fear, and poor quality of life (Dampier et al., 2010; Sil et al., 2016a). Conversely, underlying major depression as a comorbid condition may exacerbate pain and impede daily function.
“With multiple comorbidities presenting back-to-back, the sudden poor state of health takes an emotional and mental toll. I did experience anxiety and depression for several months.”
—Adult living with SCD8
“It’s really difficult to function because while the pain is affecting whichever area of your body it is, the mind starts to develop a sense of helplessness because the focus is on the pain and trying to find relief.”
—Adult living with SCD9
SUMMARY AND CONCLUSIONS
Acute and chronic pain are hallmarks of SCD and contribute to the morbidity, reduced quality of life, and disability that individuals living with the disease experience. The pathogenesis of pain in individuals with SCD is
___________________
8 Excerpt from Call for Perspectives Response.
9 Excerpt from Call for Perspectives Response.
complex and multifactorial, and worsening pain, or crisis, is not always the result of vaso-occlusion, ischemia-reperfusion injury, or nociceptive pain. Crises may also occur from a worsening of neuropathic pain, nociplastic pain, or both. People with chronic pain can experience exacerbations of their chronic pain and identify distinctly separate acute crises, which may prompt different approaches to pain management. While teasing apart these patterns helps to inform decision making about pain management, the distinctions between acute, acute-on-chronic, and chronic pain may be irrelevant to the individuals experiencing the pain. Ultimately it is the individual’s experience of pain, regardless of type, and its effect on their ability to function that are important. Pain of any etiology or from any condition is, by definition, a subjective experience, but traditional patient-reported pain scales often have limited clinical utility, given the severity of SCD-related pain and the contextual factors that contribute to pain assessment, such as interpersonal bias. Patient report is the gold standard diagnostic criterion for determining an SCD crisis.
Acute pain crises have classically been thought of as consisting of four phases, although the utility of this construct is uncertain. Research has shown that the evolution of a pain crisis is neither predictable nor linear for many individuals, particularly regarding time until resolution. Moving away from this traditionally understood structure of pain, research has determined that there is heterogeneity in the pain experienced by people living with SCD, with variability in its timing, intensity, symptoms, quality, duration, and location. Indeed, the frequency and severity of pain crises can vary widely among individuals, influenced by their unique genetic, environmental, and psychosocial factors.
People with SCD often describe their pain as throbbing, sharp, or stabbing; it can affect any part of the body but commonly affects the back, chest, abdomen, and extremities. Given the variation in experiences of pain among people with SCD, complications are variable. Repeated acute nociceptive input from vaso-occlusion can lead to central sensitization, or hyperexcitability of the central nervous system, and central nervous system inflammation, leading to long-term alterations in pain processing. This combination of recurrent acute pain episodes and central sensitization can result in a transition from acute to chronic pain in people with SCD.
Individuals living with SCD experience significant functional effects from pain. A 2020 survey conducted to assess the overall disease burden of SCD showed that those who completed the survey reported experiencing more pain crises than the literature states, with 41 percent of them reporting that acute pain crises affected their family and social life. Chronic pain from SCD can have major effects on daily functioning, health care utilization, and behavioral health, and children often experience moderate-to-severe levels of functional disability, recurrent school absences, depression, fear,
and poor quality of life from chronic pain. Many of these effects continue into adulthood and may worsen.
Based on its review of the literature, the committee reached the following conclusions:
Conclusion 2-1: Pain is a hallmark of sickle cell disease (SCD), and there is growing recognition that the majority of individuals with SCD suffer daily or persistent pain (e.g., chronic pain), as well as acute pain crises.
Conclusion 2-2: Pain severity can only be measured through self-report, since there is no validated objective measure for pain in SCD. Everyone experiences and responds differently to both chronic pain and acute pain crises.
Conclusion 2-3: The causes, frequency, severity, and duration of acute pain crises vary among individuals with SCD.
The frequency of acute care utilization for pain underestimates the true burden of pain in individuals with SCD and is too restrictive a proxy for quantifying the burden of an individual’s pain.10 Because the majority of SCD pain is treated at home, the length of an individual’s hospitalization for pain does not reflect the length of the crisis itself. Moreover, many individuals may seek to gain control of the pain in the emergency department or during a short admission and continue pain mitigation at home, so specifying at least 48 hours inside the hospital as a criterion of pain crisis severity is not clinically meaningful. In addition, studies have found that about 20 percent of people with SCD have new crises within one week of a prior crisis and 50 to 60 percent have recurrence within one month. Thus, a separation of 30 days between crises is an arbitrary cutoff for a new crisis, is not scientifically justified, and may lead to an underestimate of the impact of pain on an individual with SCD and may lead to worsened care.
Based on its review of the literature and its expert assessment, the committee reached the following conclusion:
Conclusion 2-4: The interval between discrete acute complications of SCD that may require emergency department visits or hospitalization is not predictable, and multiple discrete events may occur within 30 days.
- The requirement for a 30-day separation between emergency department visits and hospitalizations to be counted as separate events is not valid.
___________________
10 This sentence was changed after release of the report to clarify the limitations of the current proxy.
- For example, a new pain crisis, stroke, and acute chest syndrome are discrete events that could all occur within 30 days, each requiring separate emergency department visits or hospitalization.
REFERENCES
Albo, C., S. Kumar, M. Pope, K. M. Kidwell, H. Xu, L. Bowman, L. Wells, N. Barrett, S. Fields, P. Bora, N. Patel, and A. Kutlar. 2020. Characteristics and potential biomarkers of adult sickle cell patients with chronic pain. European Journal of Haematology 105(4):419–425.
Anie, K. A., and J. Green. 2015. Psychological therapies for sickle cell disease and pain. Cochrane Database of Systematic Reviews 2015(5).
Ansari, J., and F. N. E. Gavins. 2019. Ischemia-reperfusion injury in sickle cell disease: From basics to therapeutics. The American Journal of Pathology 189(4):706–718.
Bakshi, N., I. Lukombo, H. Shnol, I. Belfer, and L. Krishnamurti. 2017. Psychological characteristics and pain frequency are associated with experimental pain sensitivity in pediatric patients with sickle cell disease. The Journal of Pain 18(10):1216–1228.
Ballas, S. K., and D. S. Darbari. 2020. Review/overview of pain in sickle cell disease. Complementary Therapies in Medicine 49:102327.
Ballas, S. K., K. Gupta, and P. Adams-Graves. 2012. Sickle cell pain: A critical reappraisal. Blood 120(18):3647–3656.
Borhade, M. B., P. Patel, and N. P. Kondamudi. 2024. Sickle cell crisis. In StatPearls (Internet). Treasure Island, FL: StatPearls Publishing.
Brandow, A. M., R. A. Farley, and J. A. Panepinto. 2014. Neuropathic pain in patients with sickle cell disease. Pediatric Blood & Cancer 61(3):512–517.
Brandow, A. M., K. J. Zappia, and C. L. Stucky. 2017. Sickle cell disease: A natural model of acute and chronic pain. Pain 158(1):S79–S84.
Campbell, C. M., G. Moscou-Jackson, C. P. Carroll, K. Kiley, C. Haywood, Jr., S. Lanzkron, M. Hand, R. R. Edwards, and J. A. Haythornthwaite. 2016. An evaluation of central sensitization in patients with sickle cell disease. The Journal of Pain 17(5):617–627.
Chacko, P., E. H. Kraut, J. Zweier, C. Hitchcock, and S. V. Raman. 2013. Myocardial infarction in sickle cell disease: Use of translational imaging to diagnose an under-recognized problem. Journal of Cardiovascular Translational Research 6(5):752–761.
Citero, V. de A., J. L. Levenson, D. K. McClish, V. E. Bovbjerg, P. L. Cole, B. A. Dahman, L. T. Penberthy, I. P. Aisiku, S. D. Roseff, and W. R. Smith. 2007. The role of catastrophizing in sickle cell disease—The PiSCES project. Pain 133(1-3):39–46.
Coleman, B., H. Ellis-Caird, J. McGowan, and M. J. Benjamin. 2016. How sickle cell disease patients experience, understand and explain their pain: An interpretative phenomenological analysis study. British Journal of Health Psychology 21(1):190–203.
Collins, P. J., A. Renedo, and C. A. Marston. 2022. Communicating and understanding pain: Limitations of pain scales for patients with sickle cell disorder and other painful conditions. Journal of Health Psychology 27(1):103–118.
Dampier, C., S. Lieff, P. LeBeau, S. Rhee, M. McMurray, Z. Rogers, K. Smith-Whitley, and W. Wang. 2010. Health-related quality of life in children with sickle cell disease: A report from the Comprehensive Sickle Cell Centers Clinical Trial Consortium. Pediatric Blood & Cancer 55(3):485–494.
Dampier, C., T. M. Palermo, D. S. Darbari, K. Hassell, W. Smith, and W. Zempsky. 2017. AAPT diagnostic criteria for chronic sickle cell disease pain. The Journal of Pain 18(5):490–498.
Desai, N. J., A. L. Gonzalez-Herrera, S. Malay, T. Brown, A. Owusu-Ansah, and S. Ahuja. 2022. Influence of weather patterns and changes on sickle cell disease vaso-occlusive episodes and acute chest syndrome: A nationwide sample analysis. Blood 140(Supplement 1): 8282–8283.
Diggs, L. W., and R. E. Ching. 1934. Pathology of sickle cell anemia. Southern Medical Journal 27(10):839–845.
Douglas, S. D. 2016. Substance P and sickle cell disease—A marker for pain and novel therapeutic approaches. British Journal of Haematology 175(2):187–188.
Field, J. J., S. K. Ballas, C. M. Campbell, L. E. Crosby, C. Dampier, D. S. Darbari, D. K. McClish, W. R. Smith, and W. T. Zempsky. 2019. AAAPT diagnostic criteria for acute sickle cell disease pain. Journal of Pain Research 20(7):746–759.
Finan, P. H., C. P. Carroll, G. Moscou-Jackson, M. O. Martel, C. M. Campbell, A. Pressman, J. M. Smyth, J.-M. Tremblay, S. M. Lanzkron, and J. A. Haythornthwaite. 2018. Daily opioid use fluctuates as a function of pain, catastrophizing, and affect in patients with sickle cell disease: An electronic daily diary analysis. The Journal of Pain 19(1):46–56.
Fingar, K. R., P. L. Owens, L. D. Reid, K. B. Mistry, and M. L. Barrett. 2019. Characteristics of inpatient hospital stays involving sickle cell disease, 2000–2016. HCUP Statistical Brief #251. Rockville, MD: Agency for Healthcare Research and Quality. www.hcup-us.ahrq.gov/reports/statbriefs/sb251-Sickle-Cell-Disease-Stays-2016.pdf (accessed April 29, 2025).
Gil, K. M., K. K. Anthony, J. W. Carson, R. Redding-Lallinger, C. W. Daeschner, and R. E. Ware. 2001. Daily coping practice predicts treatment effects in children with sickle cell disease. Journal of Pediatric Psychology 26(3):163–173.
Glaros, A., and A. M. Brandow. 2020. Neuropathic pain in sickle cell disease: Measurement and management. Hematology, American Society of Hematology Education Program 2020(1):553–561.
Gordon, R. D. A., R. L. Welkie, N. Quaye, J. S. Hankins, A. A. Kassim, A. A. Thompson, M. Treadwell, C. J. Lin, and R. M. Cronin. 2024. Burden of employment loss and absenteeism in adults and caregivers of children with sickle cell disease. Blood Advances 8(5):1143–1150.
Hawkins, D. 2017. How rap revolutionary Prodigy, dead at 42, overcame the pain of sickle cell anemia. The Washington Post, June 21. https://www.washingtonpost.com/news/morning-mix/wp/2017/06/21/how-rap-revolutionary-prodigy-dead-at-42-overcame-the-pain-of-sickle-cell-anemia/ (accessed April 9, 2025).
Haywood, C., M. Diener-West, J. Strouse, C. P. Carroll, S. Bediako, S. Lanzkron, J. Haythornthwaite, G. Onojobi, and M. C. Beach. 2014. Perceived discrimination in health care is associated with a greater burden of pain in sickle cell disease. Journal of Pain and Symptom Management 48(5):934–943.
Jagtiani, A., E. Chou, S. E. Gillespie, K. Liu, L. Krishnamurti, D. McClish, W. R. Smith, and N. Bakshi. 2024. High-impact chronic pain in sickle cell disease: Insights from the Pain in Sickle Cell Epidemiology Study (PiSCES). Pain 165(10):2364–2369.
Jang, T., M. Poplawska, E. Cimpeanu, G. Mo, D. Dutta, and S. H. Lim. 2021. Vaso-occlusive crisis in sickle cell disease: A vicious cycle of secondary events. Journal of Translational Medicine 19(1):397.
Jenerette, C. M., B. J. Pierre-Louis, N. Matthie, and Y. Girardeau. 2015. Nurses’ attitudes toward patients with sickle cell disease: A worksite comparison. Pain Management Nursing 16(3): 173–181.
Jonassaint, C. R., V. L. Jones, S. Leong, and G. M. Frierson. 2016. A systematic review of the association between depression and health care utilization in children and adults with sickle cell disease. British Journal of Haematology 174(1):136–147.
Kavanagh, P. L., T. A. Fasipe, and T. Wun. 2022. Sickle cell disease: A review. JAMA 328(1):57–68.
Kissi, A., D. M. L. Van Ryckeghem, P. Mende-Siedlecki, A. Hirsh, and T. Vervoort. 2022. Racial disparities in observers’ attention to and estimations of others’ pain. Pain 163(4):745–752.
Knisely, M. R., N. Pugh, B. Kroner, R. Masese, V. Gordeuk, A. A. King, S. M. Smith, J. G. Gurney, R. Adams, T. Wun, A. Snyder, J. Glassberg, N. Shah, M. Treadwell, and Sickle Cell Disease Implementation Consortium. 2020. Patient-reported outcomes in sickle cell disease and association with clinical and psychosocial factors: Report from the Sickle Cell Disease Implementation Consortium. American Journal of Hematology 95(9):1066–1074.
Kuisell, C., R. Ploutz-Snyder, D. A. Williams, T. Voepel-Lewis, R. J. Hutchinson, K. M. Dudding, C. Bridges, and E. M. Lavoie Smith. 2023. Adolescents and young adults with sickle cell disease: Nociplastic pain and pain catastrophizing as predictors of pain interference and opioid consumption. Clinical Journal of Pain 39(7):326–333.
Lanzkron, S., J. Little, J. Field, J. R. Shows, H. Wang, R. Seufert, J. Brooks, R. Varadhan, C. Haywood, Jr., M. Saheed, C. Y. Huang, B. Griffin, S. Frymark, A. Piehet, D. Robertson, M. Proudford, A. Kincaid, C. Green, L. Burgess, M. Wallace, and J. Segal. 2018. Increased acute care utilization in a prospective cohort of adults with sickle cell disease. Blood Advances 2(18):2412–2417.
Levenson, J. L., D. K. McClish, B. A. Dahman, V. E. Bovbjerg, V. de A. Citero, L. T. Penberthy, I. P. Aisiku, J. D. Roberts, S. D. Roseff, and W. R. Smith. 2008. Depression and anxiety in adults with sickle cell disease: The PiSCES project. Psychosomatic Medicine 70(2):192–196.
Liu, S. A., T. R. Brown, A. A. King, L. A. Lin, S. S. Rehman, R. A. Grucza, and K. Y. Xu. 2024. Opioid-related emergency admissions in people with opioid dependence/use disorder with and without sickle cell disease: An analysis of multi-state insurance claims. General Hospital Psychiatry 91:83–88.
Manwani, D., and P. S. Frenette. 2013. Vaso-occlusion in sickle cell disease: Pathophysiology and novel targeted therapies. Blood 122(24):3892–3898.
Matthie, N., C. Jenerette, A. Gibson, S. Paul, M. Higgins, and L. Krishnamurti. 2020. Prevalence and predictors of chronic pain intensity and disability among adults with sickle cell disease. Health Psychology Open 7(1):2055102920917250.
Morikis, V. A., A. A. Hernandez, J. L. Magnani, M. Sperandio, and S. I. Simon. 2021. Targeting neutrophil adhesive events to address vaso-occlusive crisis in sickle cell patients. Frontiers in Immunology 12:663886.
Murray, N., and A. May. 1988. Painful crises in sickle cell disease—Patients’ perspectives. The BMJ 297(6646):452–454.
Nelson, S. C., and H. W. Hackman. 2013. Race matters: Perceptions of race and racism in a sickle cell center. Pediatric Blood & Cancer 60(3):451–454.
Niscola, P., F. Sorrentino, L. Scaramucci, P. De Fabritiis, and P. Cianciulli. 2009. Pain syndromes in sickle cell disease: An update. Pain Medicine 10(3):470–480.
Novelli, E. M., and M. T. Gladwin. 2016. Crises in sickle cell disease. Chest 149(4):1082–1093.
Osunkwo, I., B. Andemariam, C. P. Minniti, B. P. D. Inusa, F. El Rassi, B. Francis-Gibson, A. Nero, C. Trimnell, M. R. Abboud, J. B. Arlet, R. Colombatti, M. de Montalembert, S. Jain, W. Jastaniah, E. Nur, M. Pita, L. DeBonnett, N. Ramscar, T. Bailey, O. Rajkovic-Hooley, and J. James. 2021. Impact of sickle cell disease on patients’ daily lives, symptoms reported, and disease management strategies: Results from the international Sickle Cell World Assessment Survey (SWAY). American Journal of Hematology 96(4):404–417.
Osunkwo, I., J. James, F. El-Rassi, A. Nero, C. P. Minniti, C. Trimnell, J. Paulose, N. Ramscar, T. Bailey, O. Rajkovic-Hooley, and B. Andemariam. 2022. Burden of disease, treatment utilization, and the impact on education and employment in patients with sickle cell disease: A comparative analysis of high- and low- to middle-income countries for the international Sickle Cell World Assessment Survey. American Journal of Hematology 97(8):1055–1064.
Padhee, S., A. Alambo, T. Banerjee, A. Subramaniam, D. M. Abrams, G. K. Nave, Jr., and N. Shah. 2021. Pain intensity assessment in sickle cell disease patients using vital signs during hospital visits. Pattern Recognition (2021) 12662:77–85.
Pas, R., K. Ickmans, S. Van Oosterwijck, K. Van der Cruyssen, A. Foubert, L. Leysen, J. Nijs, and M. Meeus. 2018. Hyperexcitability of the central nervous system in children with chronic pain: A systematic review. Pain Medicine 19(12):2504–2514.
Phillips, S., A. M. Schlenz, S. D’Alton, M. Johnson, and J. Kanter. 2023. Patient and family opioid decision-making for pain management in sickle cell disease: A qualitative study. The Journal of Pain 24(7):1240–1250.
Platt, O. S., B. D. Thorington, D. J. Brambilla, P. F. Milner, W. F. Rosse, E. Vichinsky, and T. R. Kinney. 1991. Pain in sickle cell disease. Rates and risk factors. New England Journal of Medicine 325(1):11–16.
Power-Hays, A., and P. T. McGann. 2020. When actions speak louder than words—Racism and sickle cell disease. New England Journal of Medicine 383(20):1902–1903.
Reich, J., M. A. Cantrell, and S. C. Smeltzer. 2023. An integrative review: The evolution of provider knowledge, attitudes, perceptions and perceived barriers to caring for patients with sickle cell disease 1970-now. Journal of Pediatric Hematology/Oncology Nursing 40(1):43–64.
Ruta, N. S., and S. K. Ballas. 2016. The opioid drug epidemic and sickle cell disease: Guilt by association. Pain Medicine 17(10):1793–1798.
Schneider, M. B., A. Manikowski, L. Cohen, C. Dampier, and S. Sil. 2022. The distinct longitudinal impact of pain catastrophizing on pain interference among youth living with sickle cell disease and chronic pain. Journal of Behavioral Medicine 45(4):622–631.
Shah, N., M. Bhor, L. Xie, J. Paulose, and H. Yuce. 2019. Sickle cell disease complications: Prevalence and resource utilization. PLoS One 14(7):e0214355.
Shapiro, B. S., L. J. Benjamin, R. Payne, and G. Heidrich. 1997. Sickle cell-related pain: Perceptions of medical practitioners. Journal of Pain and Symptom Management 14(3):168–174.
Sharma, D., and A. M. Brandow. 2020. Neuropathic pain in individuals with sickle cell disease. Neuroscience Letters 714:134445.
Sigalla, J., N. Duparc Alegria, E. Le Roux, A. Toumazi, A.-F. Thiollier, L. Holvoet, M. Benkerrou, S. Dugue, and B. Koehl. 2021. Neuropathic pain in children with sickle cell disease: The hidden side of the vaso-occlusive crisis. Children 8(2):84.
Sil, S., L. L. Cohen, and C. Dampier. 2016a. Psychosocial and functional outcomes in youth with chronic sickle cell pain. Clinical Journal of Pain 32(6):527–533.
Sil, S., C. Dampier, and L. L. Cohen. 2016b. Pediatric sickle cell disease and parent and child catastrophizing. The Journal of Pain 17(9):963–971.
Sil, S., J. L. Lee, J. Klosky, A. Vaz, L. Mee, S. Cochran, B. Thompson, and R. Coakley. 2021. The comfort ability program for adolescents with sickle cell pain: Evaluating feasibility and acceptability of an inpatient group-based clinical implementation. Pediatric Blood & Cancer 68(6):e29013.
Smith, J., P. Flowers, and M. Larkin. 2009. Interpretative phenomenological analysis: Theory, methods and research. London, UK: Sage.
Smith, W. R., L. T. Penberthy, V. E. Bovbjerg, D. K. McClish, J. D. Roberts, B. Dahman, I. P. Aisiku, J. L. Levenson, and S. D. Roseff. 2008. Daily assessment of pain in adults with sickle cell disease. Annals of Internal Medicine 148(2):94–101.
Sundd, P., M. T. Gladwin, and E. M. Novelli. 2019. Pathophysiology of sickle cell disease. Annual Review of Pathology: Mechanisms of Disease 14:263–292.
Takaoka, K., A. C. Cyril, S. Jinesh, and R. Radhakrishnan. 2021. Mechanisms of pain in sickle cell disease. British Journal of Pain 15(2):213–220.
Waldrop, R. D., and C. Mandry. 1995. Health professional perceptions of opioid dependence among patients with pain. American Journal of Emergency Medicine 13(5):529–531.
White, M. K., C. Saucier, M. Bailey, D. D’Alessio, A. Foster, D. St. Pierre, and K. Raymond. 2021. Content validation of a self-report daily diary in patients with sickle cell disease. Journal of Patient-Reported Outcomes 5(1):63.
Wideman, T. H., R. R. Edwards, D. M. Walton, M. O. Martel, A. Hudon, and D. A. Seminowicz. 2019. The multimodal assessment model of pain: A novel framework for further integrating the subjective pain experience within research and practice. Clinical Journal of Pain 35(3):212–221.
Yale, S. H., N. Nagib, and T. Guthrie. 2000. Approach to the vaso-occlusive crisis in adults with sickle cell disease. American Family Physician 61(5):1349–1356, 1363–1364.

















