Genes, Behavior, and the Social Environment: Moving Beyond the Nature/Nurture Debate (2006)
Chapter: 8 Study Design and Analysis for Assessment of Interactions
8
Study Design and Analysis for Assessment of Interactions
A clear formulation of the concept of “interaction” and an understanding of the research designs that can be used to test for it are central to progress in assessing the impact of interactions among social, behavioral, and genetic factors on health. This chapter discusses definitions of interaction, from both statistical and biological points of view. It also describes the models of interaction that are likely to be relevant for evaluating the joint influence of multiple factors on health. Finally, the chapter considers research methods for the detection and elucidation of interactive effects and statistical issues related to the application of these methods.
DEFINITIONS OF INTERACTIONS
Statistical tests for interaction are entirely dependent on the measurement scale—additive or multiplicative—that is used to evaluate the effects of different factors on health. This problem is absolutely critical to the design of future studies and to the interpretation of their results. Researchers using different scales would reach different conclusions about the same underlying biological processes and make different public health recommendations depending on the scale being used. Moreover, the same data can be made to fit more than one statistical model (e.g., by analysis of the original measurement versus log-transformed data), making it very difficult to interpret the results. Thus, if the goal is to understand biology, a different way to conceptualize interaction must be found that is not dependent on statistical models. Epidemiologists have struggled with this problem and
have developed an alternative conceptual framework for interaction that is not based on statistical models (Rothman and Greenland, 1998a; Rothman and Greenland, 2005). The logic of this new conceptualization is described in detail in the paper provided in Appendix E by Sharon Schwartz and summarized below; it leads to the conclusion that the additive scale is the only meaningful reference point for the measurement of interaction. Few investigators are aware of this new conceptualization of interaction, and hence few have used it as a basis for interpreting their findings.
Statistical Interaction
From a statistical point of view, interaction can be defined as a deviation from conditional independence, a state in which the effect of one factor (social, behavioral, or genetic) on health is the same within strata defined by another factor. This definition implies that an interaction is present if the effect of a social or behavioral factor on disease risk differs among individuals with different genotypes, or if the effect of a genotype on disease risk differs among individuals with different levels of a social or behavioral factor. The problem with this definition, as indicated above, is that it is entirely dependent on the measurement scale (multiplicative or additive). Ratio measures such as relative risks (RRs) or odds ratios (ORs) assess the effects of risk factors on a multiplicative scale, because they reflect the degree to which disease risk (for RR) or odds (for OR) are multiplied in individuals with the risk factor compared to those without. In contrast, risk differences (RD) assess the effects of risk factors on an additive scale, because they reflect how much disease risk is added in individuals who have the risk factor, compared with those who do not. The statistical definition of interaction differs depending on which of these measurement scales is used. For example, in the consideration of factors A and B, interaction on a multiplicative scale is defined as a different RR for factor A across strata defined by factor B, while on an additive scale, interaction is defined as a different RD for factor A across strata defined by factor B. Use of these two different measurement scales can lead to substantively different conclusions in studies of interaction.
Table 8-1 illustrates the relationship, in general, between interaction defined on the multiplicative and additive scales. The risks to four categories of individuals are considered: those who have both a risk-influencing genotype and an environmental exposure (r11), the genotype but not the exposure (r01), the exposure but not the genotype (r10), and neither (r00). On the multiplicative scale, interaction is defined by (r11/r01) ≠ (r10/r00), while on the additive scale it is defined by (r11−r01) ≠ (r10−r00). To facilitate comparison of these two measures, it is convenient to express both as RRs, with the risk in individuals with neither genotype nor exposure as the
TABLE 8-1 Epidemiologic Measures of the Effects of a High-Risk Genotype and a Social or Behavioral Risk Factor
|
|
High-Risk Genotype |
Low-Risk Genotype |
||
|
Disease Status |
Social or behavioral risk factor present |
Social or behavioral risk factor absent |
Social or behavioral risk factor present |
Social or behavioral risk factor absent |
|
Cohort Study |
|
|
|
|
|
Affected |
a |
B |
e |
f |
|
Unaffected |
c |
D |
g |
h |
|
Risk |
r11 = a/(a + c) |
r01 = b/(b + d) |
r10 = e/(e + g) |
r00 = f/(f + h) |
|
Relative Risk |
RR11 = r11/r00 |
RR01 = r01/r00 |
RR10 = r10/r00 |
RR00 = 1.0 (referent) |
|
Case-Control |
|
|
|
|
|
Study |
|
|
|
|
|
Cases |
a |
B |
e |
f |
|
Controls |
c |
D |
g |
h |
|
Odds Ratio |
OR11 = a h/cf |
OR01 = bh/df |
OR10 = eh/gf |
OR00 = 1.0 (referent) |
reference category, so that RR11 = (r11/r00), RR10 = (r10/r00), RR01 = (r01/r00), and RR00 = (r00/r00) = 1.0. Table 8-1 illustrates how these RRs would be calculated in a cohort study and how ORs would be calculated in a case-control study, as estimates of these RRs. With these definitions it is easy to show, by simple algebra, that interaction measured on a multiplicative scale is defined by RR11 ≠ RR10 × RR01, while interaction measured on an additive scale is defined by RR11 ≠ RR10 + RR01 − 1. Thus, under a multiplicative scale, interaction would be tested for by determining whether or not RR11 is equal to the product of RR10 and RR01, while under an additive scale, interaction would be tested for by determining whether or not RR11 is equal to the sum of these two RRs − 1. The only circumstances under which the same conclusion would be drawn about whether or not interaction is present from both measurement scales is when either RR10 or RR01 is equal to 1.0—that is, one (or both) of the risk factors has no effect when acting in the absence of the other. If each risk factor increases risk when acting in the absence of the other (i.e., RR10 > 1.0 and RR01 > 1.0), the RR11 that must be observed to declare that interaction is present will be higher under a multiplicative scale than under an additive scale, thus, if a multiplicative scale is used, interaction on an additive scale could be missed. Also, if both factors have effects when acting by themselves and no interaction is present on a multiplicative scale, interaction will always be present on an additive scale.
The importance of considering the underlying model against which gene-environment interactions are tested (i.e., additive versus multiplica-
tive) is illustrated by a study of the Factor V Leiden variant, use of oral contraceptives (OCs), and risk of deep vein thrombosis (DVT) (Vandenbroucke et al., 1994; Austin and Schwartz, 2006). The use of OCs is associated with an approximately four-fold increased risk of DVT among women who do not carry Factor V Leiden (RR10 = 3.7); and, similarly, carrying Factor V Leiden is associated with a four-fold increased risk among women who do not take OCs (RR01 = 4.0). The risk of DVT in women who carry Factor V Leiden and use OCs is close to what would be expected under a model of no interaction on a multiplicative scale—that is, RR11 = 19.8, which is very similar to 3.7 × 4.0 = 14.8. However, the absolute incidence of DVT is higher among OC users compared to OC nonusers; hence, the same relative increase in risk of DVT associated with OC use in Factor V Leiden carriers and noncarriers translates into a larger absolute risk difference due to OC use among Factor V Leiden carriers—that is, RR11 (19.8) is almost three times greater than RR01 + RR10 − 1 (6.7). In short, the use of a multiplicative scale to test for interaction fails to identify an important interaction between these characteristics: excess DVT cases occur among women who are both OC users and Factor V Leiden carriers compared to what would be expected based on the sum of the individual effects of these factors (Austin and Schwartz, 2006). Such information could potentially be important, for example, in counseling Factor V Leiden carriers regarding the risks and benefits of choosing OCs for birth control as opposed to other available methods.
Newer Conceptualization in Epidemiology
To build a conceptual framework more closely tied to biology, epidemiologists begin by considering what happens at the individual level rather than at the level of the population. Much of this thinking is based on the counterfactual model, which defines a “cause” of disease, in an individual, as any factor without which the disease would not have occurred (Greenland and Robins, 1986; Rothman and Greenland, 1998b; Maldonado and Greenland, 2002; Rothman and Greenland, 2005). This framework assumes that multiple etiologic pathways (“sufficient causes”) can lead to the same disease, and within each etiologic pathway, multiple factors can work in tandem (multiple “component causes” within a sufficient cause) to cause the disease. An interaction, then, is defined as the co-participation of two component causes within one sufficient cause, so that both factors are necessary for the sufficient cause to occur. The commissioned paper by Sharon Schwartz (see Appendix E) describes this “sufficient-component cause” model in detail, specifically as it applies to the assessment of gene-environment interactions. An abbreviated description appears here.
For an individual who is exposed to a set of risk factors, only two
outcomes are possible with regard to disease occurrence: the individual either develops disease or remains unaffected. Individuals can be classified into “response types” depending on whether or not they develop disease under different exposure combinations. In the simplest case of two dichotomous risk factors, there are four possible risk factor combinations (i.e., in the case considered here: both genotype and exposure, genotype alone, exposure alone, and neither). For each risk factor combination, an individual can be either a responder (i.e., he/she develops disease) or a nonresponder (i.e., he/she remains unaffected), which means that there are 2 × 2 × 2 × 2 = 16 possible response types. For example, in one response type, individuals develop disease under all four exposure combinations (so-called Doomed). At the opposite extreme, some individuals never develop disease regardless of their genotype or exposure status (so-called Immune) (Rothman and Greenland, 1998a).
Among the 16 possible response types, 10 can be viewed as involving interaction, in the sense that an individual’s response to one risk factor depends on his/her status with respect to the other. (This definition may appear similar to the statistical concept discussed above—a deviation from conditional independence—but it is totally different because it considers the response at an individual level, rather than at the level of population measures of risk.) For example, in one interaction type, an individual develops disease only if exposed to both factors (causal synergism), and in another, an individual develops disease if exposed to either factor alone, but not if exposed to both or neither (causal antagonism). All other possible interaction types are also considered, involving either risk-raising or protective effects and either synergism or antagonism.
This model, based on conceptualizing risks at an individual level, can be used to derive an expected pattern of risk in a population under different exposure conditions. To do this, the population is assumed to contain a distribution of the different response types. The average risk, or incidence proportion, among individuals with each exposure combination is computed by adding the proportions of response types who will be affected if they have that exposure combination. When the average risks are computed in this way, under the assumption that none of the 10 interaction types is present in the population, the results show that the risks are consistent with risk additivity (i.e., no interaction on an additive scale, or (r11 − r01) = (r10 − r00)) (Rothman and Greenland, 1998a). This derivation demonstrates, based on a conceptual rather than a statistical argument, that tests for the presence of interaction should be based on risks measured on an additive scale. One important caveat should be noted in this regard, however: although departures from risk additivity reflect the presence of interaction types in the population, a lack of departure from additivity does not necessarily imply absence of interaction types. This is because different types of inter-
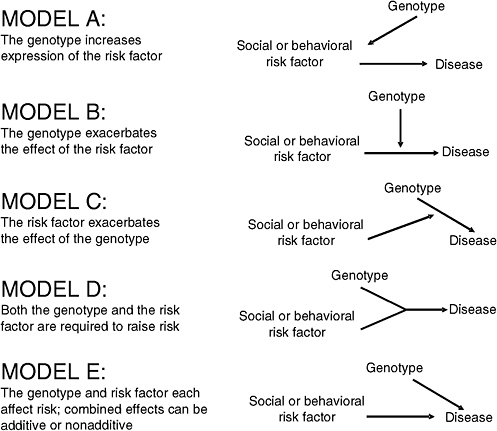
FIGURE 8-1 Five plausible models of interaction between a genotype and a social or behavioral risk factor.
SOURCE: Adapted from Ottman, 1996.
action could counterbalance one another. Moreover, departures from additivity may be difficult to detect because of limitations in statistical power. Also, most commonly used statistical software tests for interaction only on a multiplicative scale—for example, through the inclusion of an interaction term in a logistic regression model.
Plausible Models of Gene-Environment Interaction
Khoury et al. (1988) and Ottman (1990, 1996) have outlined several plausible models of interaction that are relevant for the consideration of the joint effects of the interaction of social, behavioral, and genetic factors on health (Figure 8-1). The following describes these models in both conceptual terms and in terms of the average risks expected in a population.
In model A, a genotype has a causal effect on a social or behavioral risk factor and through this pathway influences disease risk indirectly (Ottman, 1990; Ottman, 1996). This model differs markedly from the others because it does not involve interaction in either the statistical or biological sense. One hypothetical example would be a genetic influence on the behavior of alcohol consumption, which would be expected to affect risk for cirrhosis of the liver and other alcohol-related diseases. In this model a behavioral risk factor, alcohol consumption, is an intervening or mediating variable in the relation of a genotype to health. Although this model does not involve interaction, it is important for the consideration of the joint effects of genetic, social, and behavioral factors on health. An understanding of the intervening factors relevant to the effects of genes on health may facilitate the development of methods to prevent adverse health outcomes associated with genetic effects. For example, dietary treatment is used to prevent mental retardation in individuals with phenylketonuria, an autosomal recessive condition characterized by a deficiency of the enzyme needed to convert phenylalanine to tyrosine. Removal of phenylalanine from the diet prevents the buildup of blood levels of phenylalanine (the intervening variable), which would otherwise have a toxic effect on brain development. In the alcohol consumption example above, programs targeted to drinking behavior in individuals with a genetic susceptibility could reduce the risk of liver disease.
Model B postulates that a social or behavioral factor has a direct causal relationship to disease, and a genotype exacerbates this relationship without having any effect on disease when acting by itself. Returning to the example of alcohol drinking behavior, this model might involve a genetic influence on alcohol metabolism that leads to exacerbation of the health effects of drinking behavior. No effect of the genotype would be expected in the absence of exposure to drinking. In model C, a genotype is assumed to have a direct effect on disease risk, and the social or behavioral factor exacerbates this effect, without influencing disease risk when acting by itself. An example might involve a genetic influence on liver disease that is exacerbated by even small amounts of drinking (i.e., levels that do not influence risk in persons who do not carry the high-risk genotype). In model D, both a genotype and a social or behavioral risk factor are required to influence risk—neither affects risk in the absence of the other. Such a model might involve an alcohol-sensitivity genotype, heavy drinking, and liver disease—the assumption being that only individuals who drink heavily and who have a genetically mediated sensitivity to the effects of drinking develop disease. Finally, in model E both a genotype and a social or behavioral factor influence disease risk in the absence of the other factor. Although models B, C, and D involve interaction regardless of the scale of measurement, in model E the joint effect
of the two factors can be consistent with interaction on an additive scale, a multiplicative scale, or neither.
The models discussed above do not explicitly incorporate protective effects of genes and social or behavioral factors, nor antagonistic rather than synergistic interactions. These are important and are likely to become increasingly so as new chemotherapeutic interventions and pharmacogenomic applications are developed. For example, one model not included in models B, C, D, and E involves a protective effect observed only in persons with a particular genotype. This type of effect was observed in a recent study, in which postmenopausal estrogen usage was protective for cognitive impairment only in women who did not carry ApoE-ε4 alleles (Yaffe et al., 2000). A risk-raising effect of carrying ≥ 1 ApoE-ε4 allele (RR01 > 1) and a protective effect of menopausal estrogen usage (RR10 < 1) were observed, and the RR in women with both the genotype and the exposure (RR11) was greater than expected under an additive model, since the protective effect of the exposure was restricted to those without the high-risk genotype. Taken together, all of these models illustrate the potential complexity of evaluating interactions, even for a single genotype and a single social or behavioral factor.
RESEARCH DESIGNS FOR EVALUATING INTERACTIONS
A number of research designs for testing models of gene-environment interaction have been described (Ottman, 1994; Andrieu and Goldstein, 1998; Andrieu et al., 2001; Liu et al., 2004; Lake and Laird, 2004; Tweel and Schipper, 2004; Andrieu and Goldstein, 2004; Kraft and Hunter, 2005; Hunter, 2005; Moffit et al., 2005). These include traditional cohort and case-control designs with measured genotypes, sibling pair and case-parent triad designs, twin studies, and other approaches. The different designs offer advantages and disadvantages with respect to validity and efficiency. Population stratification, a special type of confounding in allelic association studies (Wacholder et al., 2002; Thomas and Witte, 2002), also may affect studies of gene-environment interaction, possibly leading to spurious findings. In allelic association studies, this type of confounding consists of a spurious association between an allele and disease, resulting from the existence of subgroups that vary both in allele frequency and disease occurrence. In a population “stratified” in this way, the distribution of subgroups will differ for affected and unaffected individuals, leading to a difference between the cases and controls in allele frequencies when the allele does not have a true association with disease. The importance of population stratification in studies of allelic association is controversial, but one recent study found evidence for substantial confounding that would not have been detected by standard analytical methods (Campbell et al., 2005). Campbell and colleagues (2005) found that a single nucleotide polymorphism (SNP)
in the LCT gene was strongly associated with height in a sample of European Americans. This association was due to stratification: both height and the frequency of the SNP varied widely across Europe. When subjects were rematched on the basis of location of European ancestry, the apparent association was greatly diminished. Since population subgroups also may be expected to differ with respect to environmental exposures, the same type of confounding could occur in studies of gene-environment interaction. That is, different subgroups may have both different genetic backgrounds and different cultures or socioeconomically influenced patterns of behavior, creating a correlation between genotype and environmental exposure that must be controlled for. Thus the impact of stratification on different designs should be considered.
Cohort Studies
Unlike retrospective case-control studies, prospective cohort studies are immune to recall bias (i.e., different recall of exposures by cases and controls) and are expected to have minimal selection bias if the follow-up rate is high. If participation in the study is unrelated to ethnicity, the susceptibility of these types of studies to confounding due to population stratification also should be minimal. However, if this is a concern, genomic control methods can be used to control for stratification effects (Pritchard and Donnelly, 2001; Devlin et al., 2001; Tang et al., 2005). Genomic control methods make use of information on unlinked genetic markers throughout the genome to assess population stratification and adjust for it, if necessary. The rationale is that with population stratification, affected and unaffected individuals are likely to differ with respect to allele frequencies at many loci throughout the genome, while if an allele is truly related to disease risk, the association is likely to be restricted to a single genomic region.
In nested case-control studies, cases and controls are ascertained from within a cohort study. That is, cases are identified at follow-up and generally are matched on appropriate covariates to a group of controls, but risk factor data collected at baseline are used as predictors. Thus, again, information on exposures is collected prior to disease onset, and the problem of recall bias is avoided. However, some selection bias (i.e., differential inclusion of some subgroups of cases, because of differential survival or loss to follow-up) may occur (although usually less than in retrospective case-control studies). The main disadvantage of these studies is the large number of subjects that must be enrolled to ensure an adequate number of cases during the follow-up period for analysis. Also, for late-onset disorders, a very long follow-up period may be needed. Prospective studies of gene-environment interaction are feasible only for common disorders, and even then, collaborative studies are essential to obtain sample sizes for sufficient statistical power.
Case-Control Studies
In retrospective case-control studies, the potential for recall bias and selection bias is considerable (Kraft and Hunter, 2005). Case-control studies also are susceptible to bias due to population stratification; this should be minimized by matching on ethnicity (Wacholder et al., 2000; Wacholder et al., 2002; Cardon and Palmer, 2003; Reiner et al., 2005), and/or using genomic control methods in the analysis (Pritchard and Donnelly, 2001; Devlin et al., 2001; Tang et al., 2005). Temporality may be difficult to establish with respect to the environmental exposures; that is, it may be difficult to ensure that exposure occurred before the onset of disease. For example, an association with a behavioral factor could be due to a change in behavior in cases following the onset of disease symptoms. Liu et al. (2004) compared the traditional case-control design with several alternatives recently suggested for testing gene-environment interaction, including the case-only (described below), partial case-control, and case-parent trio designs. They found that the validity of these alternative designs was reduced by common problems such as population stratification, genotyping error, and correlation between genotype and exposure in the population. Thus, despite the potential for bias in the traditional case-control design, they concluded it was preferable to the alternatives they assessed.
Case-Only Design
In the case-only design, interaction is assessed by testing for an association between the genotype and the exposure within the cases only (Khoury and Flanders, 1996). Such an association within the cases provides evidence for interaction because it is not expected if there is no interaction on a multiplicative scale, assuming that the genotype and environmental exposure occur independently in the population (i.e., individuals are no more likely to have both genotype and exposure than would be expected based on their individual frequencies). This design has been criticized on several grounds (Gatto et al., 2004; Hunter, 2005). First, it cannot be used to assess the main effects of a genetic or environmental factor, but only the presence or absence of interaction. Second, the assumption of independence between genetic and environmental factors in the population can be difficult to assess (Gatto et al., 2004). Third, it is a valid test for interaction only on a multiplicative scale, and not on an additive scale (Ottman, 1996).
Family-Based Designs
Several family-based designs for evaluating gene-environment interaction have been described (Witte et al., 1999; Goldstein and Andrieu, 1999),
including case-control studies using sibling or cousin controls and designs involving cases and their unaffected parents. Recently, Andrieu and Goldstein (2004) described an alternative design using both related and unrelated controls. An important advantage of family-based designs is the elimination of the potential for population stratification (although power for the detection of genetic effects is lower than in designs using unrelated controls) (Risch and Teng, 1998; Teng and Risch, 1999). Gauderman (2002) showed that designs using cases and their unaffected siblings were more efficient for the detection of interaction than were those using unrelated controls. In some studies, family members may be more willing to participate than unrelated subjects, leading to improved efficiency in data collection; on the other hand, enrollment of family members can be complicated and expensive and can involve difficult confidentiality issues. Also, in family-based studies the collection of information on exposure status generally is retrospective, leading to the same potential for recall bias as in case-control studies (Kraft and Hunter, 2005).
Mendelian Randomization
Mendelian randomization is another design used to examine the combined effects of genetic and nongenetic factors on disease risk (Davey Smith and Ebrahim, 2003). This design is not intended as a test of interaction; instead, it is designed to validate an association between an environmental exposure and disease risk. When a disease is found to be associated with an environmental exposure, it can be difficult to prove that the association reflects a true biological effect rather than confounding with some other factor. Mendelian randomization is a way to test such a biological relationship. If the exposure being studied is influenced by a genotype, the effect of the exposure can be studied indirectly by testing for an association of the genotype with disease. “Mendelian randomization” refers to the random assignment of the genotype according to Mendel’s laws, which is assumed to be less susceptible to confounding than is measurement of the exposure. The model tested with this approach is one in which the exposure serves as an intervening variable in the relation of a genotype to disease risk; this is the same as in model A (Figure 8-1).
For example, one study used Mendelian randomization to test the validity of its findings regarding blood pressure and C-reactive protein (CRP). The authors initially found that blood pressure was associated with CRP levels, but they suspected that this finding was due to confounding (Davey Smith et al., 2005). To evaluate the validity of the association, they examined the relationship between blood pressure and a genetic polymorphism in the human CRP gene that was strongly associated with CRP levels. Blood pressure did not vary by genotype of the CRP polymorphism, suggesting
the original association was due to confounding. Although a disease-genotype association may sometimes validate the idea that an environmental exposure raises disease risk, other design and analysis issues, such as population stratification, complicate studies aimed at detecting the genotype association. Also, the genotype might influence phenotypic traits other than the one being studied, thus its association with disease might not validate the relationship that it is intended to validate.
Human Laboratory Research
Human laboratory designs, in which interventions are tested in human subjects in a highly controlled laboratory setting, also can be utilized effectively to investigate the interacting effects of social, behavioral, and genetic factors on health. Such designs afford an opportunity for greater experimental control over environmental exposures and the use of interventions (within-subject designs) for exposure testing that increase statistical power. A hypothetical example would be an investigation of the effects of genetic variation in alcohol metabolizing enzymes (between subject factor) and exposure to a stress paradigm (e.g., public speaking or interpersonal stress compared to a low-stress task) (within subject factor) on alcohol sensitivity and ad lib alcohol intake. One recent human laboratory study conducted by Lerman et al. (2004) involved 71 smokers enrolled in a randomized controlled trial of buproprion treatment versus placebo for smoking cessation. The goal was to examine the degree to which abstinent smokers experience increased reward from food (possibly related to weight gain following smoking cessation), and the moderating effects of buproprion treatment and the Taq 1 polymorphism of the dopamine D2 receptor gene (DRD2). At two time points (before and after smoking cessation), subjects participated in a taste test to measure palatability of various foods followed by a behavioral economics evaluation of food reward. Carriers of the minor allele (A1) of the DRD2 polymorphism had significant increases in the rewarding value of food following smoking cessation that were not observed in noncarriers. Moreover, these effects were attenuated by buproprion treatment and predicted subsequent weight gain. Similar paradigms can be tested in animal and human laboratory models providing cross-species validation (Blendy et al., 2005). However, this approach generally is not feasible on a population basis.
Evaluation of Gene-Environment Interactions for Nonbinary Outcome Variables
In most classical epidemiologic designs, the outcome variable is binary. Other types of health outcomes include a continuous quantity (e.g., hyper-
tension and quantitative scale of depression), count (e.g., number of brain tumors), and occurrence of an event over time (e.g., timing of cancer or heart attack). Many of the intermediate phenotypes (endophenotypes) likely to be productive in the search for gene-environment interactions are quantitative rather than binary. For these nonbinary outcomes, the basic concept of gene-environment interaction remains the same: co-participation of a genotype and environmental exposure in the causal mechanism of the outcome of interest (Siemiatycki and Thomas, 1981; Rothman and Greenland, 1998a). In an empirical analysis, the assessment of statistical interaction depends on the type of outcome variable and the corresponding statistical model; linear regression, Poisson regression, and survival analysis are the standard statistical models for continuous, count, and survival outcomes, respectively. A study of gene-environment interaction would be similar to a study of interaction in general in the sense that a gene-environment interaction study still needs to follow all the established rules of the statistical model used. However, because an interaction study tends to involve more, and oftentimes many more, parameters, issues such as sample size/power and multiple testing would become more severe. With nonbinary outcomes as with binary outcomes, analysis of statistical interaction is scale dependent. To understand the underlying biology, a conceptual framework that is not rooted solely in statistical modeling is needed, and the modern epidemiologic framework also applies here (see Appendix E).
STATISTICAL ISSUES COMMON TO ALL RESEARCH DESIGNS
Sample Size and Power
A critical design issue is the determination of the minimum sample size required to generate sufficient statistical power for a study to be able to detect an interaction. That is, a sufficient sample size is needed to ensure that an effect that is truly present can be detected in the study. A study on interactions generally requires a substantially larger sample than a study only on a main effect. Roughly speaking, for the multiplicative model, detecting an interaction requires a sample at least four times as large as a sample required for detecting a main effect (Smith and Day, 1984). Power requirements are likely to be even greater for the detection of interaction on an additive scale (Garcia-Closas and Lubin, 1999). For studies of gene-environment interactions, the methods for calculating sample size have been developed for cases in which the outcome variable is categorical and the environmental exposure is binary, ordered categorical, or continuous (Hwang et al., 1994; Foppa and Spiegelman, 1997; Garcia-Closas et al., 1999; Garcia-Closas and Lubin, 1999) and for cases in which both the outcome variable and the exposure are continuous (Luan et al., 2001).
Yang et al. (2003) discussed the use of population attributable fraction to determine sample size for case-control studies of gene-environment interaction.
The sample size required for the detection of interaction between a genetic variant and a continuous environmental exposure on a continuous outcome is determined by the magnitude of the interaction, the allele frequency, and the strength of the association between exposure and outcome. In addition, statistical power and required sample size are highly influenced by the amount of measurement error in environmental exposures and phenotypes (Wong et al., 2003). Wong et al. (2003) evaluated the effect of measurement error on the sample size needed to detect gene-environment interactions. The model examined was a simple linear regression relating a continuous exposure to a continuous outcome, where the ratio of the slopes of two genotypes served as the interaction parameter. These authors found that the sample size required for the detection of interaction under this model is determined by the magnitude of the interaction, the allele frequency, and the strength of the association between exposure and outcome in those with the common allele. They also found that statistical power and required sample size are highly influenced by the amount of measurement error in environmental exposures and phenotypes, so that studies using imprecise exposure and outcome variables need much larger samples than those that utilize repeated and more precise measures. This result suggests that investment in more precise measures may be a more cost-effective approach to studies of gene-environment interaction than investment in larger samples. Vineis proposed a greatly increased investment in validated exposure assessment procedures, including incorporating repeated measures, assessment of regression dilution bias (i.e., reduction of the differences between comparison groups because of measurement errors), and validation of novel research methods (Vineis, 2004).
Pooling samples from a number of studies may be an effective means of increasing power for studies on gene-environment interaction studies in prospective analyses. The National Cancer Institute Breast and Prostate Cancer and Hormone-Related Cohort Consortium, for example, is combining data across 10 prospective studies to examine gene-environment interactions. The combined samples consist of 6,000 breast cancer patients and 8,000 prostate cancer patients based on more than 800,000 individuals and more than 7 million years of life (Hunter, 2005). An additional advantage is that these kinds of joint efforts will facilitate prior coordination and provide relatively uniform data and analyses.
Two computer programs that estimate power and sample size are available on the Internet: POWER (dceg.cancer.gov/POWER/) and QUANTO (hydra.usc.edu/gxe/).
Multiple Comparisons
The problem of multiple comparisons arises when researchers conduct several tests simultaneously and affirm a statistically significant result from any test having a p value smaller than a critical value of, say, 0.05. This approach fails to take into consideration the increased probability of a false positive test result as the number of tests performed increases. The problem is well known and commonly addressed by procedures such as Bonferroni correction, controlling the false discovery rate (FDR) (Hochberg, 1988; Benjamini et al., 2001), and the permutation test (Edgington, 1995; Nichols and Holmes, 2002).
The multiple comparisons problem poses a formidable challenge in gene-environment interaction studies, because multiple variables will be assessed, regardless of whether or not interaction is specifically evaluated. In genome-wide association scans, thousands or hundreds of thousands of SNPs are tested to search the genome for risk-associated variants. Such tests recently have been made possible by technological advances in genotyping (Thomas et al., 2005). In studies of gene-environment interactions, the multiple-comparison problem is further exacerbated. Researchers must deal not only with a large number of SNPs, but also with environmental exposures and social and behavioral factors, as well as a large number of interactions among the SNPs and the exposures. The presence of a large number of potential gene-environment interactions dramatically increases the chance of finding false-positive results if the problem is ignored. Moreover, the statistical correction for the problem, which normally requires extremely small p values, will result in a lower probability of reporting true positive interactions. That is, the possibility of not detecting important, real associations is increased in this setting.
Multiple testing must be addressed through a number of statistical procedures. The classic Bonferroni correction tends to overcorrect the problem because it assumes tests are independent when often they are not. Several advances in procedures used to correct for multiple testing have been made in recent years. One of the seminal advances was the work of Benjamini et al. (2001) who, following the ideas of Holm (1979) and Hochberg (1988), focused on controlling the FDR (defined as the percent of statistical tests that are false positives, among those deemed significant) rather than the traditional family-wise type I error rates (FEW, the probability of making at least one false positive inference). By controlling the FDR, the researchers assure themselves that on average only perhaps about 5 percent of the total positive discoveries are false. This preserves greater power to detect true positives than the more traditional Bonferroni-type FEW procedures. Several recent variations of this method have been pub-
lished (Efron and Tibshirani, 2002; Keselman et al., 2002; Sabatti et al., 2003; Storey and Tibshirani, 2003; Becker and Knapp, 2004).
Another approach uses permutation testing (Edgington, 1995; Nichols and Holmes, 2002). Here researchers take advantage of the correlation structure between the tests (testing SNPs in linkage disequilibrium will produce correlated tests of significance) in the multiple adjustment procedure. The permutation test computes significance by counting the number of ways the data can be permuted that produce results more extreme than observed (as in the Fisher’s exact test). Much less power is lost in correcting for multiple testing, since researchers are automatically accounting for the exact correlation between tests and not overcorrecting by assuming that all tests are independent.
In addition to the statistical approaches to control the false positive rate, which are largely similar to those proposed for the analysis of main effects, Hunter (2005) suggested restricting the number of interactions by biological plausibility and reproducibility. The exposures will be restricted to those that plausibly interact with genetic variants in the same biological pathways. Similarly, genetic variants will be restricted to those that plausibly alter gene function. Biological plausibility, however, is relative, and whether a particular gene-environment interaction is biologically plausible is at least partially subject to interpretation. The reproducibility of gene-environment interactions from more than one study also is crucial. The assessment of reproducibility requires the prior coordination of large studies so that the results can be compared. It is important to make all results, both positive and negative, available to avoid the publication bias of suppressing “negative” results.
CONCLUSION
This chapter has examined several aspects of the concept of interaction in order to establish a foundation for discussions about the effects of interactions among specific social, behavioral, and genetic factors in their influence on health. It has been noted that statistical definitions of interaction, and their interpretation, depend on whether the effects being studied are characterized on a multiplicative scale using relative risk measures or on an additive scale using risk difference measures. This chapter has described recent developments in epidemiology that conceptualize interaction in terms of patterns of response in individuals and that advocate the use of the additive model and has characterized a series of models that can be used to delineate the potential causal effects of combinations of multiple factors.
This chapter also considered a series of research designs that can be used to investigate these interactions. These study designs include traditional epidemiological approaches such as prospective cohort studies and
case-control studies, but also include less commonly used designs such as family studies and case-only studies. Each of these research designs has advantages and disadvantages, but all of them share common statistical challenges in the context of interactions. These challenges include obtaining sufficient study sample size to ensure that the statistical power is sufficient to detect interactions, the growing problem of multiple comparisons as genomic technologies dramatically increase the number of polymorphisms being studied, the biological plausibility of gene-disease associations, and the reproducibility of genetic association studies.
Testing for interactions will require the development of new, accessible statistical software for implementing tests for interaction on an additive scale. Furthermore, multisite collaborations may be required in order to assemble databases of sufficient size needed to ensure adequate statistical power for the testing of interactions. Additionally, several steps are needed to advance the science of testing interactions. Therefore the committee recommends the following:
Recommendation 7: Advance the Science of the Study of Interactions. Researchers should base testing for interaction on a conceptual framework rather than simply the testing of a statistical model, and they must specify the scale (e.g., additive or multiplicative) used to evaluate whether or not interactions are present. If a multiplicative scale is used, consistency with an additive relation between the effects of different factors also should be evaluated. The NIH should develop RFAs for research on developing study designs that are efficient at testing interactions, including variations in interactions over time and development.
The issues discussed in this chapter clearly illustrate the complexity of studying interactions between genetic factors and social and environmental factors and the need to consider carefully the feasibility of available research approaches. However, as we build on existing study designs and use emerging methodological and analysis techniques, it is becoming more and more possible to understand these interactions and their potential for improving public health and preventing disease on a population basis. In order to make substantive advances in this transdisciplinary work, it will be essential for genomic scientists and social scientists to learn to communicate and collaborate effectively.
REFERENCES
Andrieu N, Goldstein AM. 1998. Epidemiologic and genetic approaches in the study of gene-environment interaction: An overview of available methods. Epidemiologic Reviews 20(2):137-147.
Andrieu N, Goldstein AM. 2004. The case-combined-control design was efficient in detecting gene-environment interactions. Journal of Clinical Epidemiology 57(7):662-671.
Andrieu N, Goldstein AM, Thomas DC, Langholz B. 2001. Counter-matching in studies of gene-environment interaction: Efficiency and feasibility. American Journal of Epidemiology 153(3):265-274.
Austin M, Schwartz S. 2006. Cardiovascular disease. In: Costa L, Eaton D, editors. Gene-Environment Interactions: Fundamental of Ecogenetics. New York: John Wiley & Sons.
Becker T, Knapp M. 2004. A powerful strategy to account for multiple testing in the context of haplotype analysis. American Journal of Human Genetics 75(4):561-570.
Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. 2001. Controlling the false discovery rate in behavior genetics research. Behavioural Brain Research 125(1-2):279-284.
Blendy JA, Strasser A, Walters CL, Perkins KA, Patterson F, Berkowitz R, Lerman C. 2005. Reduced nicotine reward in obesity: Cross-comparison in human and mouse. Psychopharmacology 180(2):306-315.
Campbell CD, Ogburn EL, Lunetta KL, Lyon HN, Freedman ML, Groop LC, Altshuler D, Ardlie KG, Hirschhorn JN. 2005. Demonstrating stratification in a European American population. Nature Genetics 37(8):868-872.
Cardon LR, Palmer LJ. 2003. Population stratification and spurious allelic association. Lancet 361(9357):598-604.
Davey Smith G, Ebrahim S. 2003. “Mendelian randomization”: Can genetic epidemiology contribute to understanding environmental determinants of disease? International Journal of Epidemiology 32(1):1-22.
Davey Smith G, Lawlor DA, Harbord R, Timpson N, Rumley A, Lowe GD, Day IN, Ebrahim S. 2005. Association of C-reactive protein with blood pressure and hypertension: Life course confounding and Mendelian randomization tests of causality. Arteriosclerosis, Thrombosis, and Vascular Biology 25(5):1051-1056.
Devlin B, Roeder K, Wasserman L. 2001. Genomic control, a new approach to genetic-based association studies. Theoretical Population Biology 60(3):155-166.
Edgington ES. 1995. Randomization Tests. 3rd edition. New York: Marcel Dekker.
Efron B, Tibshirani R. 2002. Empirical Bayes methods and false discovery rates for microarrays. Genetic Epidemiology 23(1):70-86.
Foppa I, Spiegelman D. 1997. Power and sample size calculations for case-control studies of gene-environment interactions with a polytomous exposure variable. American Journal of Epidemiology 146(7):596-604.
Garcia-Closas M, Lubin JH. 1999. Power and sample size calculations in case-control studies of gene-environment interactions: Comments on different approaches. American Journal of Epidemiology 149(8):689-692.
Garcia-Closas M, Rothman N, Lubin J. 1999. Misclassification in case-control studies of gene-environment interactions: Assessment of bias and sample size. Cancer Epidemiology, Biomarkers and Prevention 8(12):1043-1050.
Gatto NM, Campbell UB, Rundle AG, Ahsan H. 2004. Further development of the case-only design for assessing gene-environment interaction: Evaluation of and adjustment for bias. International Journal of Epidemiology 33(5):1014-1024.
Gauderman WJ. 2002. Sample size requirements for matched case-control studies of gene-environment interaction. Statistics in Medicine 21(1):35-50.
Goldstein AM, Andrieu N. 1999. Detection of interaction involving identified genes: Available study designs. Journal of the National Cancer Institute Monograph (26):49-54.
Greenland S, Robins JM. 1986. Identifiability, exchangeability, and epidemiological confounding. International Journal of Epidemiology 15(3):413-419.
Hochberg Y. 1988. A sharper Bonferroni procedure for multiple tests of significance. Biometrika 75(4):800-802.
Holm S. 1979. A simple sequentially rejective multiple test procedure. Scandinavian Journal of Statistics 6:65-70.
Hunter DJ. 2005. Gene-environment interactions in human diseases. Nature Reviews Genetics 6(4):287-298.
Hwang SJ, Beaty TH, Liang KY, Coresh J, Khoury MJ. 1994. Minimum sample size estimation to detect gene-environment interaction in case-control designs. American Journal of Epidemiology 140(11):1029-1037.
Keselman HJ, Cribbie R, Holland B. 2002. Controlling the rate of Type I error over a large set of statistical tests. British Journal of Mathematical and Statistical Psychology 55(Pt 1):27-39.
Khoury MJ, Flanders WD. 1996. Nontraditional epidemiologic approaches in the analysis of gene-environment interaction: Case-control studies with no controls! American Journal of Epidemiology 144(3):207-213.
Khoury MJ, Adams MJ Jr, Flanders WD. 1988. An epidemiologic approach to ecogenetics. American Journal of Human Genetics 42(1):89-95.
Kraft P, Hunter D. 2005. Integrating epidemiology and genetic association: The challenge of gene-environment interaction. Philosophical Transactions of the Royal Society of London. Series B: Biological Sciences 360(1460):1609-1616.
Lake SL, Laird NM. 2004. Tests of gene-environment interaction for case-parent triads with general environmental exposures. Annals of Human Genetics 68(Pt 1):55-64.
Lerman C, Berrettini W, Pinto A, Patterson F, Crystal-Mansour S, Wileyto EP, Restine SL, Leonard DG, Shields PG, Epstein LH. 2004. Changes in food reward following smoking cessation: A pharmacogenetic investigation. Psychopharmacology 174(4):571-577.
Liu X, Fallin MD, Kao WH. 2004. Genetic dissection methods: Designs used for tests of gene-environment interaction. Current Opinion in Genetics and Development 14(3):241-245.
Luan JA, Wong MY, Day NE, Wareham NJ. 2001. Sample size determination for studies of gene-environment interaction. International Journal of Epidemiology 30(5):1035-1040.
Maldonado G, Greenland S. 2002. Estimating causal effects. International Journal of Epidemiology 31(2):422-429.
Moffit TE, Caspi A, Rutter M. 2005. Strategy for investigating interactions between measured genes and measured environments. Archives of General Psychiatry 62:473-481.
Nichols TE, Holmes AP. 2002. Nonparametric permutation tests for functional neuroimaging: A primer with examples. Human Brain Mapping 15(1):1-25.
Ottman R. 1990. An epidemiologic approach to gene-environment interaction. Genetic Epidemiology 7:177-185.
Ottman R. 1994. Epidemiologic analysis of gene-environment interaction in twins. Genetic Epidemiology 11:75-86.
Ottman R. 1996. Gene-environment interaction: Definitions and study designs. Preventive Medicine 25:764-770.
Pritchard JK, Donnelly P. 2001. Case-control studies of association in structured or admixed populations. Theoretical Population Biology 60(3):227-237.
Reiner AP, Ziv E, Lind DL, Nievergelt CM, Schork NJ, Cummings SR, Phong A, Burchard EG, Harris TB, Psaty BM, Kwok PY. 2005. Population structure, admixture, and aging-related phenotypes in African American adults: The Cardiovascular Health Study. American Journal of Human Genetics 76(3):463-477.
Risch N, Teng J. 1998. The relative power of family-based and case-control designs for linkage disequilibrium studies of complex human diseases I. DNA pooling. Genome Research 8(12):1273-1288.
Rothman KJ, Greenland S. 1998a. Concepts of interaction. In: Rothman KJ, Greenland S. Modern Epidemiology. 2nd edition. Philadelphia, PA: Lippincott-Raven. Pp. 329-342.
Rothman KJ, Greenland S. 1998b. Modern Epidemiology. 2nd edition. Philadelphia, PA: Lippincott-Raven.
Rothman KJ, Greenland S. 2005. Causation and causal inference in epidemiology. American Journal of Public Health 95(Suppl 1):S144-S150.
Sabatti C, Service S, Freimer N. 2003. False discovery rate in linkage and association genome screens for complex disorders. Genetics 164(2):829-833.
Siemiatycki J, Thomas DC. 1981. Biological models and statistical interactions: An example from multistage carcinogenesis. International Journal of Epidemiology 10(4):383-387.
Smith PG, Day NE. 1984. The design of case-control studies: The influence of confounding and interaction effects. International Journal of Epidemiology 13(3):356-365.
Storey JD, Tibshirani R. 2003. Statistical significance for genomewide studies. Proceedings of the National Academy of Sciences of the United States of America 100(16):9440-9445.
Tang H, Quertermous T, Rodriguez B, Kardia SLR, Zhu X, Brown A, Pankow JS, Province MA, Hunt SC, Boerwinkle E, Schork NJ, Risch NJ. 2005. Genetic structure, self-identified race/ethnicity, and confounding in case-control association studies. American Journal of Human Genetics 76(2):268-275.
Teng J, Risch N. 1999. The relative power of family-based and case-control designs for linkage disequilibrium studies of complex human diseases. II. Individual genotyping. Genome Research 9(3):234-241.
Thomas DC, Witte JS. 2002. Point: Population stratification: A problem for case-control studies of candidate-gene associations? Cancer Epidemiology, Biomarkers and Prevention 11(6): 505-512.
Thomas DC, Haile RW, Duggan D. 2005. Recent developments in genomewide association scans: A workshop summary and review. American Journal of Human Genetics 77(3): 337-345.
Tweel I, Schipper M. 2004. Sequential tests for gene-environment interactions in matched case-control studies. Statistics in Medicine 23(24):3755-3771.
Vandenbroucke JP, Koster T, Briet E, Reitsma PH, Bertina RM, Rosendaal FR. 1994. Increased risk of venous thrombosis in oral-contraceptive users who are carriers of factor V Leiden mutation. Lancet 344(8935):1453-1457.
Vineis P. 2004. A self-fulfilling prophecy: Are we underestimating the role of the environment in gene-environment interaction research? International Journal of Epidemiology 33(5): 945-946.
Wacholder S, Rothman N, Caporaso N. 2000. Population stratification in epidemiologic studies of common genetic variants and cancer: Quantification of bias. Journal of the National Cancer Institute 92(14):1151-1158.
Wacholder S, Rothman N, Caporaso N. 2002. Counterpoint: Bias from population stratification is not a major threat to the validity of conclusions from epidemiological studies of common polymorphisms and cancer. Cancer Epidemiology, Biomarkers and Prevention 11(6):513-520.
Witte JS, Gauderman WJ, Thomas DC. 1999. Asymptotic bias and efficiency in case-control studies of candidate genes and gene-environment interactions: Basic family designs. American Journal of Epidemiology 149(8):693-705.
Wong MY, Day NE, Luan JA, Chan KP, Wareham NJ. 2003. The detection of gene-environment interaction for continuous traits: Should we deal with measurement error by bigger studies or better measurement? International Journal of Epidemiology 32(1):51-57.
Yaffe K, Haan M, Byers A, Tangen C, Kuller L. 2000. Estrogen use, APOE, and cognitive decline: Evidence of gene-environment interaction. Neurology 54(10):1949-1954.
Yang Q, Khoury MJ, Friedman JM, Flanders WD. 2003. On the use of population attributable fraction to determine sample size for case-control studies of gene-environment interaction. Epidemiology 14(2):161-167.