The Great Brain Debate: Nature or Nurture? (2004)
Chapter: Part II—The Adult Brain 4 Teaching Older Dogs New Tricks
4
TEACHING OLDER DOGS NEW TRICKS
The previous two chapters emphasized the point, certainly correct, that the young developing brain is much more plastic than the adult brain. Indeed, in language and birdsong development as well as sound localization in owls, puberty or sexual maturation seems to be the point when critical developmental abilities are lost. And it is common experience that motor skills—riding a bicycle or even swinging a golf club—are much more easily learned as a youngster before puberty. And once these skills are learned as a youngster they tend to stay with us for the rest of our lives.
The view that the brain becomes quite hard-wired once we become adults is a common one, but not a correct one, and recent research on the mammalian cortex has shown that it is considerably more modifiable in adults than anyone believed just a few decades ago. The beginning of this chapter focuses on these findings, then goes on to describe some of the neurobiological mechanisms that underlie cortical plasticity—in particular, what is happening in the brain when we learn and remember things.
Plasticity of the Mammalian Cortex
The notion that the adult brain is quite hard-wired goes back at least a century. Santiago Ramón y Cajal, the great Spanish neuroanatomist who many believe is the father of modern neuroscience, wrote in 1913 in the conclusion of his work on Degeneration and Regeneration of the Nervous System: “In adult centers the nerve paths are something fixed, ended, immutable.” However, studies in several cortical areas indicate that significant modifications in cortical structure and function can occur in adults. A number of these relate to changes in response to cortical damage, but others are in response to more normal experiences. Clearly, we can learn and remember new things all our lives, and the cortex is involved in learning and memory, as we shall see. But for many decades this was thought to be a special exception, that most of the adult mammalian brain was “immutable” as Cajal suggested.
Hints that this view is not correct came first perhaps from psychological experiments, which showed that if you place ocular prisms on human beings so that the world they see is upside down, the subjects adapt within a few days and then respond to visual stimuli quite normally thereafter. When the prisms are removed, again the subjects compensate, usually very quickly (in about a day) and they again respond quite normally to visual stimuli.
This result is in stark contrast to experiments on frogs in which their optic nerves are first severed, and then the eyes rotated 180° in the head. In cold-blooded vertebrates, the optic nerve regenerates and the axons grow back to make synapses on the neurons they originally contacted. Following regeneration of the optic nerves, these animals responded exactly as if their visual world was upside down, which it was after their eyes were rotated. When feeding, they misdirected their movements by 180°: When a fly appeared in the upper right quadrant of their visual field, they reacted with a movement toward the lower left quadrant, and this aberrant behavior was permanent. The frogs never recovered from it. Thus, cold-blooded vertebrates do seem to have
a much more hard-wired nervous system than mammals. Their nervous systems have other features distinct from those of mammals as well—for example, an ability to regenerate central nervous system axons. We shall return to this topic in the next chapter.
The psychological experiments using prisms on human subjects did not teach us anything about the underlying cortical mechanisms involved or even if their compensation was cortical in nature. The first evidence for structural modifications as a result of altered sensory input to the cortex came from studies carried out by Michael Merzenich and his colleagues at the University of California, San Francisco. Using monkeys, they studied how sensory input from the fingers is first processed and represented on the cortex. Somatosensory information, representing touch, pressure, temperature, and pain from all over the body surface, is first processed in the cortex along a cortical strip, called the primary somatosensory area, located just behind the primary motor area.
The surface of the body is represented on this area in an orderly and consistent way, although the body representation is not strictly proportional. This is shown in Figure 4.1, a drawing based on the studies of Wilder Penfield, a Canadian neurosurgeon who electrically stimulated the human brain during operations for epilepsy. When the primary somatosensory area was stimulated, the patients reported a sensory sensation from a specific part of the body. Those parts of the body where sensation is more acute have more nerve endings, which in turn occupy more cortical area. Thus, the face and hand take up more cortical area than other parts of the body. The same is true for the primary motor area; electrical stimuli there caused a particular part of the body to move. A greater area of the primary motor cortex is concerned with those parts of the body that we can move more precisely, such as the fingers and parts of the face like the lips, mouth, and jaw. Undoubtedly, this larger cortical representation relates to the greater dexterity and sensory acuity of the hands and face compared to other parts of the body.
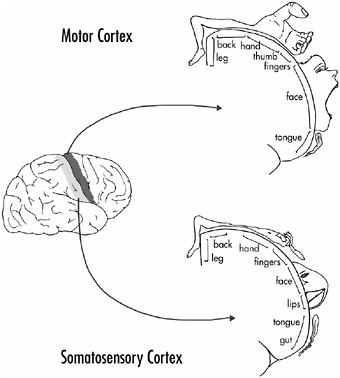
FIGURE 4-1The primary somatosensory and motor areas of the primate (human) cerebral cortex. The two drawings to the right show the body part associated with each area, as indicated. The body representations are not proportional; areas of the body where sensation is more acute or that exhibit finer movements (such as the hands and face) have greater cortical representation.
These cortical representations are also termed topographic maps, and the fingers are mapped on the somatosensory cortex so that each provides sensory input to a specific region of the cortex. These regions are sequentially arranged as shown in Figure 4-2A.
By recording from individual neurons in the hand/finger region of the somatosensory cortex and determining which finger is giving a particular neuron its sensory input, Merzenich and his colleagues first found that monkeys vary substantially in how much representation their fingers have on the cortex. Some monkeys have more cortical representation for a particular finger or groups of fingers than others. But of more interest was their finding that if the sensory nerves coming from a finger are cut (called
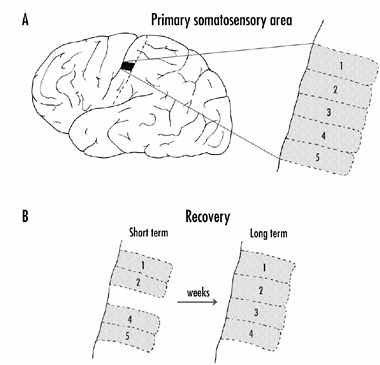
FIGURE 4-2A: Representation of the digits on the primary somatosensory area of the monkey cortex.
B: Reorganization of the cortex following severing of the sensory nerves coming from one finger (digit 3). Initially, the area of the cortex from the deafferentiated finger was silent, but with time the area received input from neurons coming from adjacent fingers. The remaining fingers then had an increased representation on the cortex.
deafferentation), or an entire finger was removed, the representation of the fingers on the cortex changed quite dramatically. Initially, when they recorded from neurons in the area that received input from the lost or deafferentated finger, the neurons were silent as shown in Figure 4-2B. Stimulation of any finger or part of the hand produced no activation in most of the neurons. The exceptions were some neurons on the edges of the area in question, which probably shared some innervation with adjacent fingers, although this input was normally silent (a topic to which we shall return).
With time, however, it was possible to activate all the neurons in the deafferentated part of the cortex by stimulating adja-
cent fingers or, in some cases, other parts of the hand. This took time—weeks, even months—but the adjacent fingers gradually increased their representation and filled in the silent area. The adjacent digits now had a larger representation on the cortex than before as shown in Figure 4-2B. The conclusion from these experiments seems inescapable: New synapses and, presumably, new neuronal branches, can be formed in the adult cortex.
A question arising from these experiments is how much reorganization can take place in the adult cortex following deafferentation or loss of a part of the body. In the experiments involving the loss of a finger, the filling in of the silent cortex was relatively limited—it represented alterations in just 1-2 mm of cortex. In more extensive deafferentation experiments, carried out in monkeys by other investigators for a different purpose, the innervation to the cortex from an entire limb was cut. Eventually (the recordings were not made until 12 years after the deafferentation) the entire hand-arm region of the somatosensory cortex filled in, a distance of 10-14 mm along the cortex. Adjacent to the hand-arm region on the somatosensory cortex is innervation from the face as shown in Figure 4-1, and stimulation of the face, especially the lower jaw and chin, now activated neurons from the deafferentated area. Exactly how long it took for this reorganization of the somatosensory cortex to take place is not clear. As noted, the recordings were not made for more than a decade following the deafferentation.
Experiments by Vilayamur Ramachandran of the University of California in San Diego suggest a similar reorganization of the cortex in humans who have had a limb amputated. If the face of an arm amputee is touched lightly with a piece of cotton, the subject reports a sensation of the amputated hand being touched. Indeed, a crude representation of the hand is found on the face as shown in Figure 4-3.
Touching the cheek induces a sensation of the thumb being touched; the upper lip, stimulation of the index finger; and below the lips, touching of the little finger. It is likely that the face area has expanded into the limb area on the cortex. That the subject
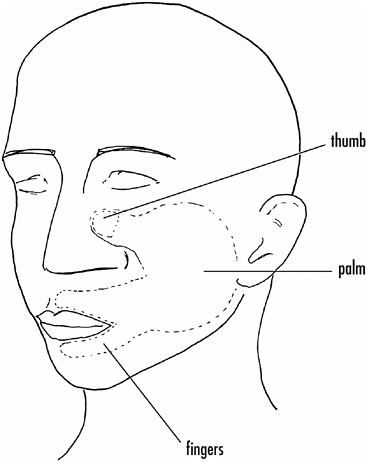
FIGURE 4-3If the face of an individual who has lost an arm is touched lightly with cotton, the individual often reports the sensation of the missing hand being touched. The face of such individuals carries a crude sensory representation of the hand.
experiences limb sensations following light touching of the face is of enormous interest. It has been proposed that this might relate to “phantom pain,” in which amputees describe sensations and even pain from amputated limbs.
Learning New Tricks
Merzenich and his colleagues also did converse experiments—to look for cortical changes following extensive stimulation of fin-
gers. These experiments showed that if monkeys were trained to use the tips of two or three fingers to rotate a disk to get food, after several thousand disk rotations over three weeks to several months, the somatosensory cortical area for the monkeys’ finger-tips had expanded. Furthermore, each cortical neuron whose activity was recorded received input from a smaller area on a particular finger, suggesting a higher touch acuity for these finger-tips. If rotation of the disk was limited to just one finger, the cortical expansion was limited to that finger.
Some other observations on these monkeys are worth noting. Following extensive and simultaneous stimulation of two or three fingers lasting several weeks, not only was the cortical area expanded for the stimulated fingers, but the areas had fused to some extent. An individual neuron recorded in the expanded area often had input from more than one finger, something never seen in normal animals.
Another most interesting finding was that the animals in these experiments lost the ability to move the affected fingers independently, indicating that changes had occurred in the motor system pathways as well. Indeed, a similar type of reorganization is observed in the primary motor cortex in monkeys trained to manipulate small objects with their fingers. The amount of motor cortex devoted to finger movement expands after training, whereas the cortical area devoted to wrist or lower arm movement contracts. If a monkey is trained to do a task involving the lower arm, the opposite result is observed—the digit cortical area is reduced, whereas the lower arm area is increased.
Do similar changes take place in humans who have practiced specialized motor tasks? The most compelling evidence comes from magnetic source imaging (magnetoencephalography or MEG) studies on the cortical representation of the left-hand “fingering” and right-hand “bowing” fingers of string, mainly violin, players. The cortical representation for the fingers of the left hand is greater than the cortical representation of the right-hand fingers in string players. As might be expected, the cortices of subjects who learned to play before age 12 showed more dramatic
increases in left-hand finger representation than those of musicians who began to play later in life. However, subjects who learned to play after age 12 still had a significantly greater representation of the left-hand fingers on the cortex compared to their right-hand fingers. Another study involving human subjects who were proficient Braille readers and from whom MEG recordings were made found that the recorded responses were significantly larger for the “reading” finger compared to the same finger on the other hand.
What about other parts of the cortex—do they show similar plastic changes to those of the somatosensory or motor cortex? The answer, gleaned from experiments on both the primary visual and auditory cortices of various mammals, is yes. If lesions are made in the corresponding parts of both retinas in a monkey, the area of primary visual cortex receiving input from that region of the visual field is initially silent. Over a period of weeks to months, the silent area begins to respond again to visual stimuli placed in adjacent regions of the visual field. Eventually, the entire area can be activated by spots of light projected onto the retinas. The same type of result has been reported in guinea pig auditory cortex. After lesions are made to the cochlea in the inner ear that destroy the animal’s ability to hear certain tones, the cortex reorganizes so that the part of the cortex once receiving input from the lesioned area is now responsive to tone frequencies sensed by adjacent areas of the cochlea.
Another obvious and important question is whether the entire adult mammalian brain exhibits plasticity or whether plasticity occurs only in the cortex. Recall that with visual deprivation during the critical period in cats and monkeys, the cortex was much more profoundly affected than was the lateral geniculate nucleus (LGN) or retina. The same holds true for adult plasticity. Whereas some plastic changes can be shown to occur in subcortical structures in adults, the major site of plasticity seems to be in the cortex. For example, Charles Gilbert and Torsten Wiesel of the Rockefeller University showed that whereas affected cortical areas respond again to retinal stimula-
tion two months after binocular retinal lesioning, the corresponding areas in the LGN remain silent.
A similar result was observed with monkeys trained to do a tactile task involving multiple digits in which the digits received simultaneous stimulation. Eventually a number of cortical neurons responded to stimulation of more than one digit, something that was not seen in control animals. However, recordings from thalamic neurons that receive input from the digits and that then project to the somatosensory cortex never showed multiple-digit input. This suggests that the convergence of sensory information from the different digits happens in the cortex.
What mechanisms underlie these cortical reorganizations? This is not well documented but, as noted above, is thought to reflect the sprouting of processes and the formation of new synapses within the cortex. The anatomical changes that take place in the visual cortex of rats exposed to an enriched environment, and described at the end of Chapter 2, might provide a model. As was described, most of these cortical changes could be induced in both young and adult rats. In confirmation of this idea, Gilbert’s laboratory has shown that after retinal lesioning, axons from cortical neurons surrounding the deafferentated area extend collateral branches into the deprived area. Axons from the LGN, on the other hand, do not expand into the deprived area; they continue to innervate only the cortical areas they originally innervated. Thus, the reorganization of the cortex appears to represent primarily cortico-cortical plasticity, and not plasticity of input to the cortex. An expansion of cortico-cortical axons could be coupled with an increase in synapses made by terminals of new axonal branches.
Mechanisms of New Synapse Formation: The Hippocampus and Memory and Learning
There is enormous interest among neuroscientists in discovering the basis for learning and memory. The notion that learning and memory involve an alteration in synapses by, for example, in-
creasing or decreasing synaptic strength or by the sprouting of new neuronal processes and formation of entirely new synapses goes back to the nineteenth century, but only recently has there been substantial evidence to back up these ideas. And, as neuroscientists have uncovered the neurobiological phenomena occurring in those parts of the cortex known to be involved in memory and learning, it has become increasingly clear that similar phenomena are occurring all over the brain, both during development and in the adult cortex.
The guess is that there are common mechanisms underlying the plasticity that happens during development as well as when we learn and remember new things. It is also likely that similar mechanisms come into play during the reorganization of the adult cortex that occurs as a result of peripheral lesioning or extensive training. Since these phenomena are best understood from studies on memory and learning mechanisms, I shall use this work as a model.
The Hippocampus and Consolidating Memories
A cortical area, termed the hippocampus, tucked underneath the temporal lobe of the cortex, has been implicated as a key structure in memory formation for at least half a century. Cortical areas adjacent to the hippocampus, particularly those that provide input to the hippocampus, are also critical for long-term memory formation. We have two hippocampi, one under each hemisphere of the brain as shown in Figure 4-4, and we would hardly notice the loss or degeneration of one hippocampus. However, if both are lost, along with adjacent cortical tissue, the result is devastating. We no longer remember things for more than a few minutes.
This happened dramatically in 1953 to a young man who had severe epilepsy, believed to originate in these regions of his brain, and who had his hippocampi and adjacent cortical areas removed neurosurgically. His epilepsy was cured, but he could no longer remember things for more than a short time. He retained most
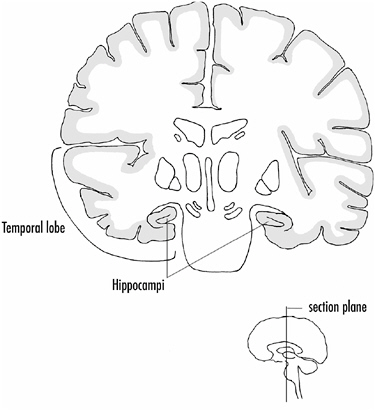
FIGURE 4-4A vertical section through the middle of the brain (insert) showing the location of hippocampi under the temporal lobes.
memories of events that had occurred before the operation, but quickly forgot new experiences or facts. He had, in other words, lost the ability to consolidate memories.
Psychologists, particularly a Canadian, Brenda Milner, have intensively studied this patient, called by his initials HM, for more than 40 years. Virtually no changes have occurred in his ability to remember new facts or events over this period, and he remembers most of them for only 20-30 seconds, what is called short-term memory. If he keeps thinking about a new fact or event, focusing his attention on it, he can continue to recall it for longer periods (a form of short-term memory called working memory), but if he becomes distracted, he quickly forgets it. His short-term memory—the ability to remember things for seconds to min-
utes—appears unimpaired. What is missing is an ability to remember things long term—for more than a few minutes.
In the course of her study of HM, Milner discovered that he could learn new motor skills, which suggests that not everything we learn depends on the hippocampus. Indeed, learning new motor skills appears to involve another, noncortical, part of the brain, the cerebellum. However, what we know about mechanisms underlying cerebellar motor learning suggests that they are similar to those happening in the hippocampus.
Long-Term Potentiation in the Hippocampus
The hippocampus, like structures elsewhere in the brain, has a highly distinctive cellular organization, having three major cell types: granule cells, CA3 pyramidal cells, and CA1 pyramidal cells. Input to the hippocampus activates the granule cells, which in turn activate the CA3 pyramidal cells. These then activate the CA1 pyramidal cells whose axons provide the main output of the hippocampus. Thus, the hippocampus has a relatively simple cellular organization, shown schematically in Figure 4-5A.
In the early 1970s, Timothy Bliss and Terje Lømo, who were working in London, made a striking observation that began an explosion of research on the hippocampus and hippocampal cells that continues to this day. While recording from one of the hippocampal neurons, they found that if you provide a strong activating stimulus to the axons providing input to the hippocampus, the subsequent response of the neuron to a weak stimulus is dramatically increased. Figure 4-5B shows this schematically. The response measured is the peak voltage change that occurs in the neuron as a result of activating the synapses onto the cell with a weak stimulus. In other words, the strong or potentiating stimulus makes the synapses onto the cell more powerful; their efficacy is increased substantially. This phenomenon is called long-term potentiation or LTP.
If a strong input stimulus is repeated several times over a relatively short period, the potentiation of the synapses lasts for days
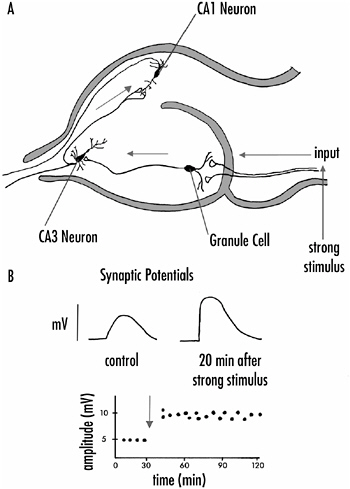
FIGURE 4-5A: The major synaptic pathways in the hippocampus. Input axons activate granule cells, which in turn synapse on CA3 neurons. Axonal branches from the CA3 neurons innervate CA1 neurons which provide the major output of the hippocampus.
B: The generation of long-term potentiation in hippocampal cells. Synaptic potentials recorded from hippocampal neurons are increased in amplitude (potentiated) for several hours following the presentation of a strong stimulus to the input axons to the hippocampus.
or even weeks. A single strong input stimulus, on the other hand, lasts for one to three hours. Thus, investigators distinguish short-term and long-term forms of LTP and these differ to some extent in their underlying mechanisms. The major point is that with repeated strong stimuli it is possible to alter a neuron’s respon-
siveness for long periods—days to weeks—and this, of course, immediately suggests how we remember things. That is, it suggests how neuronal excitation (an experience) can cause a long-term change in the nervous system (memory).
It turns out that not only can one induce LTP in many neurons, it is also possible to induce long-term depression or LTD. Following a strong input stimulus to some neurons, the synapses onto that neuron are decreased in effectiveness for short or long periods. Again, LTD is found in many neurons throughout the brain and can result in depressed synaptic activity for substantial periods.
Synaptic Mechanisms Underlying LTP
Weak stimuli do not induce LTP by themselves; they must be paired with a strong stimulus; then the weak stimuli show evidence of LTP as shown in Figure 4-5B. Neuroscientists talk of this as associative: Stimuli must be paired. LTP is associative in another way—to induce it requires activity in both presynaptic and postsynaptic cells, that is, both the cells making the synapses and the cells receiving synaptic input must be activated.
Why both presynaptic and postsynaptic cells must be active to elicit LTP is now understood. It depends on one of the receptor molecules present at synapses on the postsynaptic cell. A presynaptic axon terminal releases a chemical (neurotransmitter) from its synapses when it is activated. The neurotransmitter diffuses across a narrow bit of extracellular space, the synaptic cleft, to interact with receptor proteins on the postsynaptic side of the synapse. A presynaptic terminal is activated when the voltage across its membrane decreases—scientists say that the membrane is depolarized. The response elicited in the postsynaptic cell is also electrical at most synapses, but the postsynaptic response might be either an increase (hyperpolarization) or a decrease (depolarization) in membrane voltage. Depolarization of a postsynaptic cell is associated with excitation of the neuron—hyperpolarization with neuron inhibition, but this does not enter into our story of how LTP is generated.
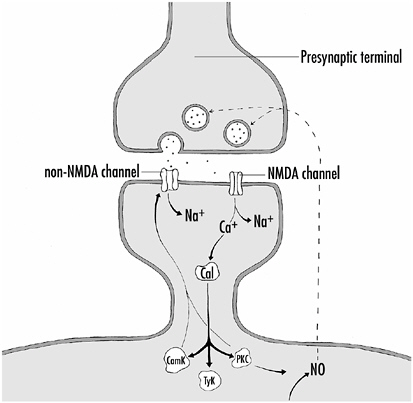
FIGURE 4-6Mechanisms establishing LTP. When the presynaptic terminal is depolarized, glutamate is released from synaptic vesicles and interacts with channels on the postsynaptic membrane. The non-NMDA channels allow Na+ into the postsynaptic cell, leading to depolarization of the cell and allowing the NMDA channels to admit both Na+ and Ca2+ into the cell. The Ca2+ binds to calmodulin (Cal), which in turn activates several kinases, some of which (CamK, PKC) phosphorylate the non-NMDA channels, increasing their effectiveness in admitting Na+ into the cell. PKC also promotes the generation of NO within the cell, which can diffuse out of the cell and into the presynaptic terminal, thereby increasing its effectiveness in releasing glutamate. Thus, LTP results from enhanced responsiveness of the non-NMDA channels to glutamate and enhanced glutamate release from the presynaptic terminal.
The neurotransmitter released at hippocampal synapses is glutamate, an amino acid. At synapses where LTP is generated, there are two types of receptor proteins in the postsynaptic membrane that interact with glutamate as shown in Figure 4-6.
Both, when activated, form channels in the membrane, allowing positively charged ions to enter the cell. Because cells are nor-
mally electrically negative inside—they have an excess of negative charges inside the cell resulting in a resting voltage or potential—the entry of the positively charged ions depolarizes the cell—the voltage across the cell’s membrane decreases.
Receptor proteins that allow ions to flow across cell membranes are called channels, and the two channel types here are NMDA channels or non-NMDA channels. NMDA (n-methyl-d-aspartate) is a chemical that specifically activates the NMDA channel; it has no effect on the non-NMDA channels, and thus can be used to differentiate the two channel types.
The non-NMDA channels are like most excitatory channel proteins found at various synapses throughout the brain. When activated by glutamate, they immediately open, allowing Na+ ions to flow into the cell and depolarize it. However, the NMDA channel works in a more complex manner, and it is key for generating LTP. If the cell is at its normal resting potential (70 mV inside negative or, conventionally, −70 mV), glutamate released from the presynaptic terminal binds to the NMDA channel, but its channel opening is blocked. It opens only if the postsynaptic cell is depolarized to some extent. The block is caused by a Mg2+ ion sitting in the entrance to the NMDA channel at resting membrane voltage.
Depolarization of the cell, which makes the inside of the cell more positive, pushes the positively charged Mg2+ ion out of the channel’s mouth, and other ions can now enter it. Here, again, the NMDA channel is different; whereas most channels allow just monovalent ions that have just one charge to flow across the membrane, that is, Na+, K+, or Cl−, the NMDA channel allows both monovalent Na+ and K+ ions and a divalent ion with two charges (Ca2+) to cross the cell membrane. And it is Ca2+ entry into the cell that is crucial for LTP, as we shall see.
But first let’s consider how the postsynaptic membrane becomes depolarized, allowing for the unblocking of the NMDA channel. This comes about by the activation of the non-NMDA channels that allow Na+ to enter the cell and depolarize it. We can now understand why activity in both the presynaptic and postsynaptic cells is required for LTP to occur and why a strong potentiat-
ing stimulus is effective in generating LTP. Such potentiating stimuli strongly activate the presynaptic cell, causing it to release substantial amounts of glutamate. The glutamate, by activating the non-NMDA channels, depolarizes the postsynaptic neuron, thereby allowing for the unblocking and activation of the NMDA channels and the entry of Ca2+ into the cell.
How does Ca2+ influx into a neuron lead to LTP? Within the neuron, Ca2+ binds to a calcium binding protein called calmodulin. When activated by Ca2+, calmodulin can activate a variety of kinases, our old friends that phosphorylate proteins and thereby alter their properties. Calmodulin can activate at least three different kinases in neurons, but how they increase the postsynaptic response is still not entirely clear. One possibility is that the kinases phosphorylate the non-NMDA channels, thereby increasing their sensitivity to glutamate, or alternatively by increasing the amount of Na+ they permit into the cell following glutamate activation. Phosphorylation of non-NMDA channels is known to do this at various synapses. This, then, is a postsynaptic mechanism.
There is evidence also for an increased release of transmitter from the presynaptic terminal during LTP—a presynaptic mechanism at play. How might this come about? One suggestion for which there is supporting evidence is that kinase activation in the postsynaptic neuron results in the generation of a messenger molecule that diffuses from the postsynaptic cell to the presynaptic terminal and increases synaptic transmitter release. The gas, nitric oxide (NO), has been implicated as this messenger molecule, and the enzymes and substrates for the production of NO are present in many neurons. Figure 4-6 shows the mechanisms for establishing LTP.
Long-Term LTP
I noted earlier that there are short-term and long-term forms of LTP. A single potentiating stimulus produces LTP lasting one to three hours, whereas four or more such stimuli produce LTP lasting for days to weeks. Short-term or early LTP (E-LTP) can be ex-
plained by the mechanisms shown in Figure 4-6, but long-term or late LTP (L-LTP) involves more elaborate pathways and more permanent changes in the cells and their synapses. For example, new protein synthesis occurs in L-LTP, but not in E-LTP.
Figure 4-7 shows schematically the mechanisms involved in L-LTP. In this process too, Ca2+-activated calmodulin is involved.
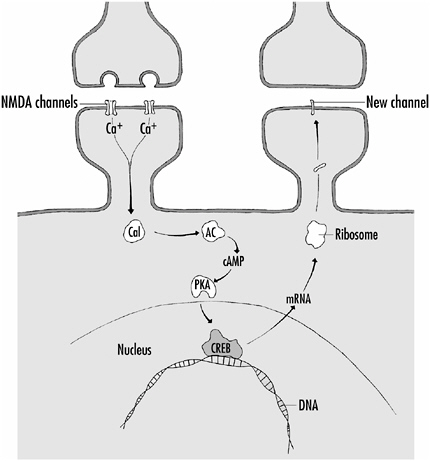
FIGURE 4-7Mechanisms underlying L-LTP. Ca2+ entering the cell via NMDA channels activates calmodulin (Cal) which in turn activates the enzyme adenylate cyclase (AC). AC catalyzes the production of the second-messenger cyclic AMP (cAMP) which then activates a kinase (PKA). PKA phosphorylates a transcription factor (CREB) which interacts with DNA in the cell nucleus leading to gene expression and the production of messenger RNA (mRNA). mRNA moves out of the nucleus and interacts with ribosomes which results in the production of new protein. The newly made proteins can, for example, make new channels that are inserted into the membrane.
Scientists believe that if sufficient calmodulin is activated, it interacts with an enzyme called adenylate cyclase (AC). This enzyme converts a molecule called adenosine triphosphate (ATP) to a smaller cyclic molecule, cyclic AMP (cyclic adenosine monophosphate). Cyclic AMP is called a second-messenger molecule because it can readily diffuse in the cell and interact with proteins. For the production of L-LTP, the cAMP interacts with a specific kinase protein (PKA) and this leads to the phosphorylation of a transcription factor called CREB (cyclic AMP response element binding protein).
Transcription factors, as described in Chapter 1, interact directly with those regions of genes in the nucleus (the promoter regions) that turn on or off gene expression. When a gene is to be turned on—that is, expressed—the code for the protein to be made is transcribed from the gene’s DNA into a piece of another, slightly different nucleic acid, RNA. The messenger RNA (mRNA) moves out from the nucleus of the cell to the cytoplasm where it is translated into protein by structures called ribosomes. In this way, new protein is made that can lead to the strengthening of synapses by, for example adding new channel proteins to them, to the formation of entirely new synapses, or even to the development of new branches made by the neuron.
LTP Elsewhere in the Cortex
LTP has been observed widely in the cortex, including in the primary visual, auditory, and motor cortices. It is usually easier to elicit LTP in the hippocampus than elsewhere in the cortex, probably because of the relatively simple and straightforward hippocampal cellular organization shown in Figure 4-5. Nevertheless LTP has been recorded in many brain structures. Furthermore, NMDA receptors have been identified throughout the cortex, so scientists believe that the mechanisms for eliciting LTP in the hippocampus probably apply elsewhere.
There is enormous interest in long-term LTP (L-LTP) in particular, because it provides a compelling model of how new neu-
ronal processes and synapses can be generated in both the young and adult brains and how synaptic circuitry can be altered in the brain as a result of experience. On the other hand, we know little as yet about the natural stimuli required to elicit LTP in various parts of the cortex and elsewhere. In the laboratory, LTP is usually generated in specific neurons by applying artificial stimuli to the axons providing input to the neurons. Some recent studies, on the other hand, show that certain auditory and visual sensory stimuli can induce LTP-like potentiation in some neurons.
A feature of LTP generation of developmental interest has been observed in the visual cortex of young rats and cats. Whereas LTP can be generated in the visual cortex of young animals, the ease of its generation is age dependent. As the animals mature, it becomes harder and harder for them to generate LTP in visual cortical neurons. This decline in LTP generation roughly parallels the critical period for the solidification of the ocular dominance columns in the cortex. The idea that the two phenomena are related is strengthened by the finding that dark-rearing, which prolongs the ocular dominance critical period, also prolongs the time it is possible to generate LTP in the visual cortex.
A similar critical period for the generation of LTP has been shown for neurons in the somatosensory cortex. Thus, LTP-like mechanisms might underlie the pruning and refinement of synapses that happen in early development, as well as the rearrangement and addition of synapses that occur both during critical periods and in the adult cortex. The bottom line is that, while the underlying mechanisms of all these phenomena are likely to be similar, their extent and ease of generation might vary with age and brain region.
Neuromodulation and Silencing Synapses
There are mechanisms by which neurons and their synapses can be modified other than through NMDA receptors and LTP. This is frequently referred to as neuromodulation and is generally thought to involve changes in neurons that last from minutes to
hours. Neuromodulation does not usually involve protein synthesis, although it can. Whereas it might take 15-20 minutes for LTP to be generated fully in a neuron, neuromodulatory effects typically begin within 15-20 seconds of their initiation.
Many of the same players involved in the various forms of LTP are also involved in neuromodulation. For example, the best-characterized neuromodulatory system involves the second messenger, cyclic AMP, and protein kinase A. Figure 4-8 shows a scheme for neuromodulation involving these substances.
At synapses where neuromodulation occurs there are receptor proteins in the postsynaptic membrane that interact with the substance released from the presynaptic terminal (a first messenger). In the case of neuromodulatory synapses, the chemical substances released, the first messengers, are most often monoamines, such as dopamine or serotonin, or small peptides, the neuropeptides. The membrane receptors they bind to don’t form channels in the membrane, but interact with one of a set of proteins called G-proteins. The G-proteins serve to activate enzymes such as adenylate cyclase, which produces second-messenger molecules such as cyclic AMP. Most often, cyclic AMP activates the PKA kinase, which then can act at virtually any level of the cell—at the membrane to alter the properties of membrane proteins including channels, in the cytoplasm to activate or inactivate enzymes, or in the nucleus to turn genes on or off by the activation of transcription factors.
We now know a variety of such neuromodulatory pathways involving the production of different second messengers, the activation of many different kinases and so forth. One of the striking phenomena that result from the activation of these pathways is the strengthening or weakening of synaptic activity. Indeed, synapses can even be silenced in this way. That is, they are present but they are not functioning or they are functioning so weakly that they are ineffective. Neural plasticity that occurs relatively rapidly in the brain—and there are many examples—probably relates more to neuromodulatory effects than to LTP and structural alterations in the nervous system.
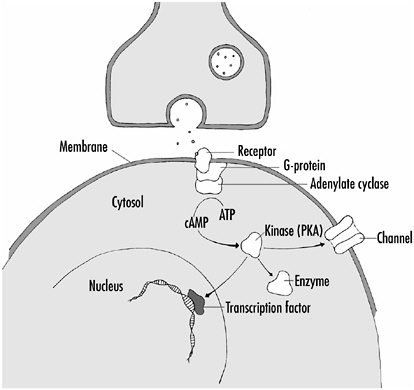
FIGURE 4-8How neuromodulation can affect a neuron. The neuromodulatory substance released from the presynaptic terminal activates a membrane protein called a receptor. When activated, the receptor protein interacts with and activates a second protein (G-protein), which in turn leads to the activation of an enzyme, in this case adenylate cyclase, which converts ATP to cyclic AMP (cAMP). The cAMP activates the kinase PKA, which then can phosphorylate proteins in the cell membrane (channels), cytosol (enzymes), or nucleus (transcription factors)—thereby activating or inactivating them and altering the properties of the neuron.
I noted earlier that after the removal of a finger from a monkey some neurons on the edges of the cortical area receiving input from that finger quickly—within minutes—give responses to stimulation of adjacent fingers. It is likely that this activity is the result of silent or weak synapses now coming into play or being greatly strengthened as a result of activation by a neuromodulatory pathway. On the other hand, it could be the result of the simple removal of inhibitory input to the adjacent finger neu-
rons that are providing this input. While it is not clear which is responsible, it is clear that a variety of mechanisms can alter synaptic strength and circuitry in the adult brain—from simple synaptic excitation and inhibition, to strengthening or weakening of synaptic strengths by neuromodulatory mechanisms, to neurons sprouting new branches and forming new synapses by mechanisms such as L-LTP.
These mechanisms can have quite different time courses, from milliseconds for ordinary excitatory and inhibitory synaptic effects, to minutes for neuromodulatory effects, to hours, days, or weeks for substantial remodeling of neurons and the formation of new synapses by mechanisms such as L-LTP. These different mechanisms are not always distinct, but represent a continuum of phenomena that generate an enormous richness of cortical plasticity and underlie a variety of mental phenomena.
I end the chapter by giving two examples of cortical plasticity that relate to perceptual phenomena. The first involves the modifications of the properties of orientation-specific neurons in the visual cortex. In Chapter 2, I described the receptive field properties of such neurons, shown in Figure 2-3A. Ordinarily these neurons respond best when a bar or edge of light with a specific orientation is present in the appropriate part of the visual field, that is, within the receptive field of these neurons. Bars or edges of light of any orientation do not activate these cells when projected onto the retina outside the receptive field. These neurons, then, appear wired to respond to a bar of light of a particular orientation in a restricted region of the visual field.
However, if several bars of light of a somewhat different orientation are placed outside the receptive field of the recorded cell and then the receptive field orientation of the neuron is reexamined, the neuron’s orientation selectivity might be altered as shown in Figure 4-9A.
The bars of light outside the receptive field do not activate the recorded cell when they are applied; their effect is to change silently the properties of the recorded cell. Not every cortical neuron shows the effect and the change in orientation selectivity appears limited—to about 10° or so. Nevertheless, it shows that even
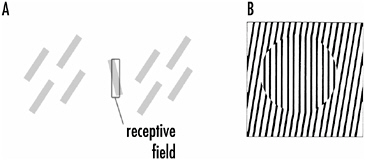
FIGURE 4-9A: The orientation tuning of a cortical neuron can be altered by up to 10° by the presence of oriented stimuli outside the receptive field. The orientation selectivity of the neuron (clear bar) is tilted to the left (shaded bar) when bars of light tilted to the right are presented outside the receptive field.
B: The tilt illusion. The lines in the circled area appear tilted to the left although they are vertical.
fundamental receptive field properties of cortical neurons such as orientation selectivity have some plasticity.
What causes these neurons to be plastic is not entirely clear, but the answer might be found in collateral branches of cortical neurons that can extend several millimeters horizontally in the cortex. Although these collateral branches have been known anatomically for some time, their function is obscure. Indeed, it might be that the synapses they make are often silent and are activated only under specific conditions such as during the experiment just described.
What consequence might such effects have? Visual physiologists have proposed that the phenomenon described above relates to the “tilt illusion,” in which the perceived orientation of a line depends on the orientation of surrounding lines, and not on the actual orientation of the lines. Figure 4-9B shows this. The lines in the middle of the figure are vertical, but because the surrounding lines tilt to the right, the central lines appear to tilt left. The more general purpose of such effects might have to do with integrating information from different parts of a complex scene—helping to explain, perhaps, the effects of context in visual perception.
Not only can peripheral stimuli outside the receptive field of
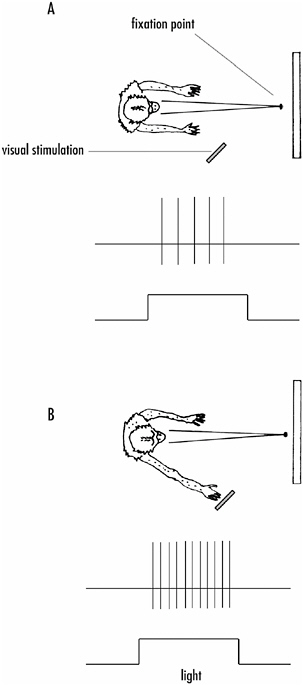
FIGURE 4-10The effect of attention on the responsiveness of a neuron in the cortex.
A: The neuron being recorded is activated by a visual stimulus in the periphery of the visual field. When the monkey is looking at the fixation point in the center of its visual field, the response of the neuron to the peripheral stimulus is minimal.
B: If the monkey attends to the peripheral stimulus by touching it, the response of the neuron is significantly enhanced, even though the monkey has not shifted its gaze from the fixation point.
a cell affect a cell’s responsiveness, but attention of the subject can also alter responsiveness. Robert Wurtz and Michael Goldberg at the National Institutes of Health first showed this. If the activity of a neuron along the visual pathways in a monkey is recorded, the strength of that activity can be increased by having the animal focus attention on the stimulus that drives the recorded neurons.
Figure 4-10 shows the experiment. A monkey is trained to keep its eyes on a fixation point in front of it. A visual stimulus then comes on elsewhere in the visual field, but within the receptive field of the recorded neuron. The visual stimulus activates the recorded neuron, but if the visual stimulus is modest in strength, so is the recorded response (in this case the action potential output of the cell, which consists of just four events).
If instead the monkey is trained to focus attention on the stimulus by reaching out and touching it when it appears (for a reward, of course), without removing its eyes from the fixation point, the activity of the recorded neurons to the stimulus is considerably enhanced.
We do not know the mechanisms underlying the plasticity illustrated by these two examples. Almost certainly they don’t involve structural changes in the brain because they occur very quickly and are rapidly reversible. However, they show how apparent rearrangement of neural circuitry and altering neuronal responsiveness can occur in the cortex, probably as a result of increasing or decreasing the effectiveness of synapses; that is, whether the synapses transmit stronger or weaker signals or even transmit signals at all.