Preventing and Treating Dementia: Research Priorities to Accelerate Progress (2025)
Chapter: 3 Understanding Disease Pathways to Guide Effective Strategies for Precision AD/ADRD Prevention and Treatment
3
Understanding Disease Pathways to Guide Effective Strategies for Precision AD/ADRD Prevention and Treatment
Dementia etiology is multifactorial, involving complex interactions between genes, exposures, human physiology, and behaviors, all of which are influenced by the aging process. The brain is a dynamic and highly adaptive organ, and its capacity to meet the cognitive demands of daily life is influenced by the cumulative effects of growth, disease, and adaptation across the life course. Of course, the brain does not operate in isolation—all systems within the body interact with and are dependent on one another to keep an organism healthy (i.e., homeostasis) (Hampel et al., 2019). For example, the endothelial cells of the vascular system maintain the blood–brain barrier that regulates the brain–body exchange, the muscular system influences the beneficial effect of exercise on the brain, and the endocrine system links peripheral metabolic status to the functioning of the brain. More recently, the effect of the gut–brain axis on the body’s—and brain’s—physiology and pathophysiology has received increasing attention (Cocean and Vodnar, 2024; Zheng et al., 2023). Thus, a healthy body is essential to maintain a healthy brain and to support healthy aging.
Interventions to effectively prevent and treat Alzheimer’s disease and related dementias (AD/ADRD) have seen limited success, in large part owing to immense gaps in our understanding of how chronic exposures (e.g., common chronic diseases such as hypertension, and social disadvantage), gene–environment interactions, and other biological pathways contribute to the different types of dementia. Moreover, while clinical symptoms of Alzheimer’s disease (AD) and those of related dementias (e.g., Lewy body, frontotemporal, vascular) may present similarly (e.g., cognitive impairment), differences in the underlying causal biology and in the disease onsets and
trajectories can be challenging to detect clinically. This poses an impediment to targeting and tailoring prevention and treatment approaches to the individual type of dementia.
Adding to this complexity is the predominance of mixed neuropathologies on autopsy, especially in older people, indicating that multiple pathological pathways are operating simultaneously and may require combinatorial interventions to achieve meaningful improvement. Consequently, different types of neurodegeneration as diagnosed clinically can show large overlaps in pathology at the brain tissue level (i.e., mixed pathology). Given the multiple and potentially intersecting pathways that contribute to AD/ADRD, diverse streams of research focused on individual pathogenetic processes and, most importantly, their integration, are needed to understand risk and resilience factors, as well as the biological processes leading to disease. The findings from such research will support the expansion of the current portfolio of prevention and treatment strategies (discussed in Chapter 4).
This chapter begins by framing the prevention and treatment of AD/ADRD in the context of healthy aging and the optimization of brain health across the life course. It goes on to explore key exposures and mechanistic pathways that may be intervened upon individually or in combination to optimize brain health over the life course and prevent and treat AD/ADRD. It focuses specifically on disease pathways that may be shared across AD/ADRD, as well as disease-agnostic (i.e., disease-nonspecific) resilience mechanisms. With this approach, the committee aims to identify opportunities to maximize the impact of AD/ADRD research investments and advocate for the transition away from the siloed study of individual causes of dementia that has dominated the research landscape and that fails to address the reality of mixed etiology dementia. The chapter ends by identifying research priorities directed at evidence gaps that, if addressed, could guide the development of population- and individual-level AD/ADRD intervention strategies.
HEALTHY AGING, RESILIENCE, AND BRAIN HEALTH ACROSS THE LIFE COURSE
Healthy aging can be defined as “a continuous process of optimizing opportunities to maintain and improve physical and mental health, independence, and quality of life throughout the life course” (PAHO/WHO, 2024). The later years of life present many new opportunities for older individuals, from pursuing new activities and long-neglected passions and even new career paths. Yet the chance to enjoy these opportunities depends to some degree on healthy aging.
As discussed further in this chapter, aging in the biological sense results from the deterioration of myriad molecular and cellular structures and
processes over time. Importantly, however, these changes do not occur on a linear trajectory and do not strictly associate with a person’s age in years (WHO, 2022). Chronological age represents the amount of time a person has been alive. Although there is no single definition for biological age, it is conceptualized in terms of various molecular and cellular changes that accumulate over time (Jazwinski and Kim, 2019). Building on the work of a 2014 trans-institute Geroscience Interest Group Summit sponsored by the National Institutes of Health,1 López-Otín and colleagues (2023) recently proposed 12 hallmarks of aging, which spanned from the molecular to the meta-organism levels (see Figure 3-1). Manipulation of these hallmarks, which are interdependent, may accelerate or decelerate the biological aging process. Many of these hallmarks overlap with the biochemical and cellular pathologic pathways common to AD/ADRD (discussed later in this chapter), suggesting opportunities to use knowledge of biological aging processes in the development of interventions for AD/ADRD (Fillit et al., 2023).
Biological age is increasingly recognized as a better indicator of chronic health outcomes as compared to chronological age (Ho et al., 2023), with implications for lifespan and brain health span (the period of life during which an individual remains healthy) (Sierra, 2016). Biological age is not necessarily linked to chronological age, given that a person can be biologically younger or older than their chronological age suggests (Sierra et al., 2021; Zhang and Gladyshev, 2020). As a result, there is substantial diversity in the range of physical and cognitive ability seen in older age. While genetics plays a role, a large part of this variability arises from the physical and social environments in which people live and the effect of these environments on their opportunities and health behaviors (i.e., social determinants of health) (WHO, 2022), as supported by increasing evidence from animal and human studies (Durso et al., 2022; Horvath and Levine, 2015; Kalia et al., 2022; Liu et al., 2023a; Quach et al., 2017; Ward-Caviness et al., 2016). Complementary to biological hallmarks of aging, social hallmarks of aging, such as low socioeconomic status, minority status, adverse life events, adverse psychological states, and adverse behaviors, can accelerate aging, while reducing exposure to these social hallmarks can slow the onset of age-related poor health outcomes (Crimmins, 2020; Hooten et al., 2022).
The risk of sporadic forms of dementia increases as individuals age, typically accompanied by age-related biological changes in the brain and throughout the body that may not be observed in individuals with early-onset forms of the diseases (Scheltens et al., 2021). Some characteristics of neurodegenerative disease (e.g., neurodegeneration and cerebral atrophy,
___________________
1 Geroscience is a transdisciplinary field focused on the relationship between biological aging and age-related diseases (Fillit et al., 2023; Hara et al., 2019).
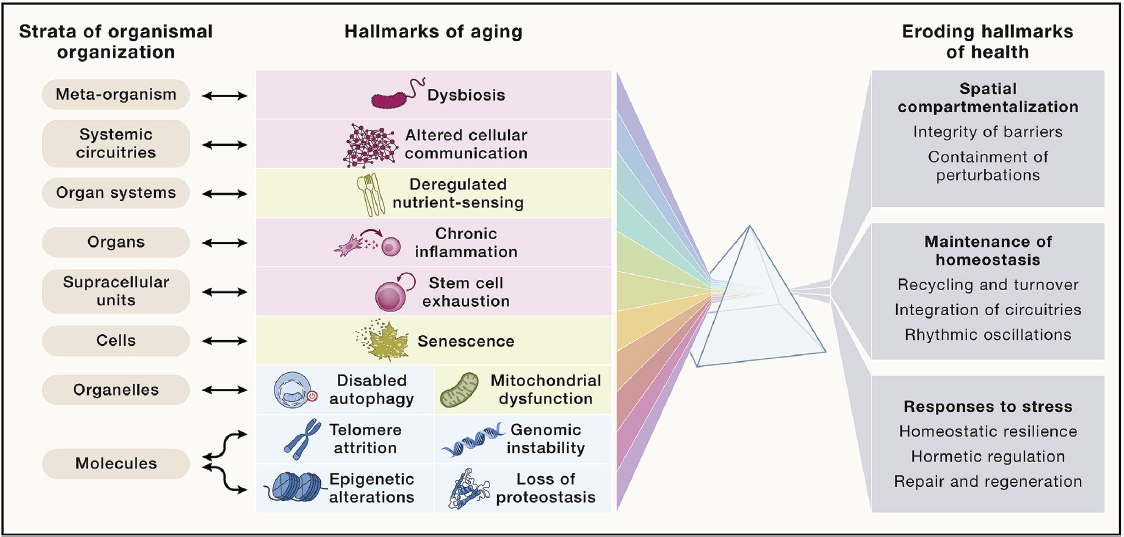
SOURCE: López-Otín et al., 2023. Reprinted with permission from Elsevier.
neuropathological protein accumulation, cognitive decline), gradually manifest over the life course even among individuals who do not develop clinical dementia (Zhang et al., 2023e). Thus, biological pathways underlying both aging and AD/ADRD exist on a continuum and are likely to overlap (Gonzales et al., 2022). Consequently, interventions targeting the underlying biological pathways associated with aging may be useful for delaying or even preventing the onset of AD/ADRD (Cummings et al., 2023a; Fillit et al., 2023; Hara et al., 2019; Sierra, 2016; Sierra et al., 2021).
It should be emphasized that the accumulation of neuropathologic changes with aging does not necessarily lead to cognitive decline. Studies to correlate neuropathologic changes with premortem cognitive function have demonstrated that cognitive health can be maintained despite the accumulation of high levels of neuropathology (Zhang et al., 2023e). This phenomenon has been defined as cognitive resilience, which refers to “the capacity of the brain to maintain cognition and function [despite the presence of] aging and disease” (Stern et al., 2023, p. 3).2 The growing understanding of cognitive resilience and the implication for potential intervention strategies is important given that people generally care more about maintaining their cognitive and functional ability than about the neurobiological changes that may occur with aging. Building cognitive reserve or enhancing the brain’s ability to compensate or adapt to neuropathology—which does not necessarily imply a return to a previous state of function but rather the ability to continue to function despite the presence of pathology—are strategies for increasing an individual’s resilience (Stern et al., 2019), and may contribute to preventing, delaying, and/or slowing clinical disease progression.
Cognitive resilience is not dichotomous but rather exists on a continuum that includes better- and worse-than-expected performance for a given level of pathology (Zammit et al., 2023). Thus, mechanisms can either maintain or degrade resilience. For example, psychosocial risk factors such as depression, loneliness, and chronic distress increase risk of clinical dementia independent of effects on typical pathologic features such as amyloid plaques, tau tangles, Lewy bodies, or infarcts (Wilson et al., 2003, 2006, 2007a,b), indicating the existence of yet-to-be discovered mechanisms that degrade resilience. Other factors such as education, cognitive stimulation, socialization, sense of purpose, and physical activity may function in a protective manner to increase or maintain resilience, enabling the brain to
___________________
2 Multiple definitions and frameworks have been proposed to clarify terminology and operationalize the concepts of resilience and resistance (Neuner et al., 2022; Stern et al., 2019, 2023; Zammit et al., 2023). For the purposes of this report, resistance to neuropathology is considered to be distinct from resilience and refers to the absence of neuropathology or lower levels than would be expected based on age and other risk factors (e.g., genetic risk) (de Vries et al., 2024; Zammit et al., 2023). This definition for resistance is similar to the definition for brain maintenance provided in one recently published consensus framework (Stern et al., 2023).
compensate for brain pathologies and maintain cognitive function (Zammit et al., 2023). As discussed later in this chapter, the discovery of protective genetic variants has shown that resilience can also be genetically mediated. Thus, there are both modifiable and nonmodifiable factors that contribute to resilience and may in fact interact. The neural mechanisms that underlie resilience are an active area of study (de Vries et al., 2024; Neuner et al., 2022). Understanding how protective variants and other mediators promote resilience may guide new strategies for interventions.
Strategies focused on avoiding gerogens—age-accelerating factors, such as pollution and stress (NASEM, 2020a,b)—and adopting resilience-promoting lifestyle factors, such as healthy diet, exercise, regular sleep patterns, and social activities, are applicable to both antiaging and AD/ADRD prevention efforts and may support the extension of the health span and the optimization of brain health across the life course. Assessment of individuals’ aging clocks using genetic, epigenetic, metabolomic, and other tools could in the future guide more personalized intervention strategies.
THE EXPOSOME AND BRAIN HEALTH ACROSS THE LIFE COURSE
Defining and Measuring the Exposome
The concept of the exposome emerged 2 decades ago and was originally described as encompassing “life-course environmental exposures (including lifestyle factors), from the prenatal period onwards” (Wild, 2005, p. 1848). In recognition of the public health importance of gene–environment interactions, the term was coined in an effort to draw attention to the need to characterize environmental exposures with the same scientific rigor and precision that was at the time being applied to the analysis of the human genome. Understanding the interplay between the genome and the environment depends on the ability to accurately assess environmental exposures (Siroux et al., 2016). Exposomics as a scientific discipline has continued to mature over the last 20 years and was recently defined as “a field that studies the comprehensive and cumulative effects of physical, chemical, biological, and psychosocial mediators that impact biological systems by integrating data from a variety of interdisciplinary methodologies and streams” in order to “enable discovery-based analysis of environmental influences on health” (Banbury Center, 2024). As suggested by this definition, exposures are considered broadly, extending well beyond environmental pollutants (e.g., air pollution, environmental lead) that may commonly be a focus of exposure assessments. Such factors as social determinants of health and health behaviors are also encompassed within the exposome.
For diseases for which genetic variants explain only a limited fraction of the variability in risk and that are presumed to be largely driven by
gene–environment and age-associated interactions (i.e., acquired susceptibility), exposomics may provide one potential approach for better understanding disease pathways. The field relies on advanced data analytics and causal inference methods to identify the relationship between exposome factors and phenotypes of interest (e.g., disease outcomes) (Banbury Center, 2024). Findings from such efforts can guide and complement effective prevention strategies.
A notable challenge, however, relates to how the exposome is defined and comprehensively measured over the entire life course given its vastness of scale and inherent variability over time, as well as how it is linked to health (Siroux et al., 2016). Exposome measurement and evaluation methods use multiple technologies and multilayered data sources, including demographic surveys, administrative data (e.g., Social Security records, health care claims), historical information (e.g., geocoded residential histories), sensors and monitors, satellite data, and analysis of endogenous and exogenous compounds in the body that are biological indicators of exposures (Banbury Center, 2024; Miller, 2024). High-resolution mass spectrometry and other increasingly high-throughput multiomics approaches (e.g., epigenomics, transcriptomics, proteomics, metabolomics) are enhancing the identification of such biological exposure indicators, as well as early molecular changes that precede disease onset (Vineis et al., 2020).
Although a data-layering approach enables the exploration of interactions among different environmental exposures over the life course and consideration of high-risk exposure windows, such large-scale data collection, management, and analysis are costly and integration of these diverse data types requires sophisticated analytic tools (Siroux et al., 2016; Vermeulen et al., 2020). Moreover, the simultaneous analysis of multiple exposures and their effects on health raises concerns regarding risk of misattribution of the effect of one exposure to another (i.e., exposure misclassification) and can make it challenging to identify causal associations because of correlations between exposures in the exposome (Siroux et al., 2016). Data security and privacy considerations must also be considered given that exposome research may involve the linking of some forms of protected health information, such as residential history and genetic information (Safarlou et al., 2023).
Linking the Exposome to Brain Health and AD/ADRD
Findings from twin studies suggest that while genetics is a major contributor to AD/ADRD risk (Gatz et al., 2006), environmental and age-related factors also have a substantial influence (Maloney and Lahiri, 2016; Migliore and Coppedè, 2022; Paulson and Igo, 2011). Yet the role of environmental factors in AD/ADRD etiologies, either directly through effects on
the brain or indirectly via altered brain–body interactions, remain poorly understood (Finch and Kulminski, 2019).
In the context of brain health, the exposome paradigm can be conceptualized in the form of the neural exposome, which encompasses a diverse array of non-genetic factors that influence neurodegenerative diseases (including AD/ADRD) and other neurological conditions (Tamiz et al., 2022). The neural exposome includes exogenous (e.g., originating from outside the organism such as environmental contaminants), behavioral (e.g., lifestyle), and endogenous factors (e.g., microbiome), as depicted in Figure 3-2. The brain’s dynamic and adaptive nature, characterized by continual change as a function of experience and exposures across the life course, makes the study of the neural exposome complex. Additionally, just as genetic variants function together to influence genetic disease risk,
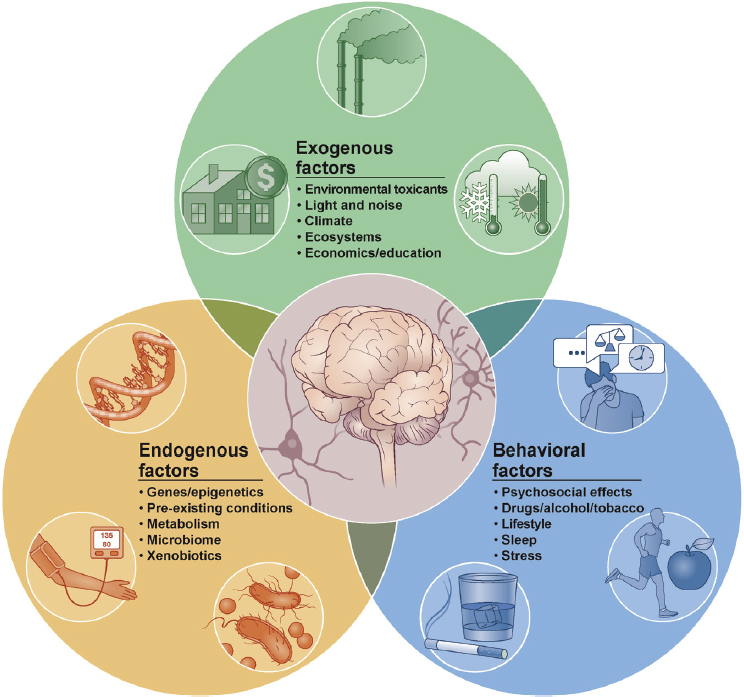
SOURCE: Tamiz et al., 2022. Reprinted with permission from Elsevier.
combined exposures from across the life course similarly are likely to have interactive effects on brain health (Tamiz et al., 2022). This complicates intervention efforts as strategies targeting a single factor may be expected to have limited effectiveness when different factors contribute additively or synergistically to the risk of AD/ADRD. Further complexity stems from the fact that exposures may be happening in early life (in so-called critical windows of development) but not manifesting until later in life. This is the basis for the developmental origins of health and disease hypothesis (Klocke and Lein, 2019; Migliore and Coppedè, 2022).
The human brain continues to develop through adolescence. Consequently, exposures (environmental and social) from gestation through teenage years can disrupt brain development in ways that will not be immediately apparent if the affected regions are not functional until later life or if compensatory mechanisms mask the effects until additional insults accumulate to a level that can no longer be compensated for. Though not related to neurodegeneration, an illustrative example is provided by the effects of monosodium glutamate, which, in high concentrations, can cause neuronal death in the hypothalamus of a developing brain. Deficits from the loss of those neurons does not become apparent until adolescence when the function of those cells is normally activated (Klocke and Lein, 2019). Whether and to what degree AD/ADRD have a developmental basis remains unclear but may be uncovered by further study of the exposome.
There has been growing investment in research on the exposome and, of particular interest to this report, its role in brain aging and disease, with the National Institute of Environmental Health Sciences (NIEHS), the National Institute on Aging (NIA), and the National Institute of Neurological Disorders and Stroke (NINDS) providing support for such research (Cavaliere and Gülöksüz, 2022; NIA, 2020; Tamiz et al., 2022). The growth of research on the exposome and AD/ADRD has created opportunities for greater interagency collaboration to advance critical research across multiple conditions. For example, in 2022, NINDS launched its Office of Neural Exposome and Toxicology, which is focused on interdisciplinary research on the neural exposome and the creation of partnerships within NINDS and across other National Institutes of Health (NIH) institutes, centers, and offices (ICOs), federal agencies (Centers for Disease Control and Prevention, Environmental Protection Agency), advocacy groups, and scientific societies (Tamiz et al., 2022). Additionally, several NIH ICOs (NIA, NINDS, NIEHS, the National Institute of Arthritis and Musculoskeletal and Skin Diseases, and the National Cancer Institute, among others) are cofunding a new coordinating center for exposome research to accelerate precision environmental health (Columbia University, 2024; NIH, 2023).
The contribution of exogenous environmental exposures to AD/ADRD is a topic of active investigation. For example, a growing body of evidence
from epidemiological and experimental studies has linked air pollution (e.g., fine particulate matter, nitrogen dioxide, nitrous oxides) and other environmental pollutants (neurotoxic metals, pesticides) to increased risk of neurologic disorders, including dementia (Bakulski et al., 2012; Liu et al., 2023a; Migliore and Coppedè, 2022; NASEM, 2020a,b; Peters et al., 2019; Shi et al., 2021; Shih et al., 2007; Wilker et al., 2023; Zhang et al., 2023a), although data are lacking for the vast majority of chemicals to which humans are exposed. There is an opportunity to leverage existing databases, emerging technologies for high-throughput screening (Lefevre-Arbogast et al., 2024), and increased understanding of the pathogenesis of AD/ADRD to identify specific environmental chemicals that interact with biological pathways involved in neurodegeneration to promote disease, and conversely, resilience.
Research is also elucidating the influence of social, cultural, and behavioral factors on brain health. Behavioral factors such as physical inactivity, smoking, alcohol consumption, and diet are well-established risk factors for dementia (Livingston et al., 2024; Patten and Lein, 2019). However, recent studies focused on social and economic factors such as educational attainment, wealth, access to health care (Chen and Zissimopoulos, 2018), loneliness and social isolation (Huang et al., 2023), macro-level social systems, and structural influences have identified associations with cognitive impairment and dementia (Lamar et al., 2023; Powell et al., 2020; Reuben et al., 2024; Walsemann et al., 2023). Such research is critical to understanding how social disadvantage and disparities in exposures to environmental hazards (e.g., neighborhood proximity to landfills and major highways that generate traffic-associated air pollution) contribute to differences in AD/ADRD risk across diverse populations. There is also increasing evidence to suggest that gender roles (e.g., household, childrearing, and caregiving roles) and disparities (e.g., access to education, occupational opportunities, autonomy) contribute to gender differences in AD/ADRD incidence (Mielke, 2024). Continuing to uncover these kinds of associations and their interactions will require longitudinal cohort studies with in-depth exposome measurements and analysis.
To understand the exposome and how it interacts with molecular and cellular mechanisms to influence AD/ADRD risk, it is critical to capture the context in which exposures occurred over an individual’s life course (e.g., socioeconomic status, locations in which individuals lived throughout their lives) (Melcher et al., 2024). Such contextual factors can significantly influence the exogenous, endogenous, and behavioral facets of the exposome, and are drivers of disparities in AD/ADRD outcomes (NASEM, 2024).
The linking of social phenotypes (e.g., social vulnerability) to biological phenotypes (e.g., neuropathology markers) can be done prospectively or retrospectively (Kind, 2024; Powell et al., 2020). As an example of the
latter, public data tracing methods can be used to collect life-course residential histories for individuals whose brains were donated to Alzheimer’s Disease Research Center brain banks, thereby linking historic population-level exposure data to existing biobank samples (Melcher et al., 2024). Retrospective approaches, however, are resource intensive and subject to the availability of accurate data. Collection of social phenotype data in parallel to biological data may be less costly and less labor-intensive. The Health and Retirement Study has been collecting social, economic, and demographic data on a nationally representative sample of middle-age and older Americans since 1992, as well as biomarker data on the study respondents (Biomarker Network, 2024). A structural and social determinants of health module has been developed and is being implemented in Alzheimer’s Disease Research Centers to enable the linking of prospectively collected social determinants of health data to neuropathology findings, which will help improve understanding of how social determinants of health modify cognition and AD/ADRD risk (Stites et al., 2022). As noted earlier in this chapter, however, privacy laws and regulations and data security concerns related to protected health information can impede efforts to conduct such linking of studies. Research infrastructure addressing both administrative and legal challenges is needed to facilitate the intake and linkage processes for protected health information (Kind, 2023).
The findings from exposome studies suggest opportunities for individual-level, public health, and policy interventions aimed at primary prevention, such as reducing exposure to environmental contaminants, modifying lifestyle, and eliminating structural factors that contribute to social disadvantage. It is possible to imagine precision prevention approaches that stratify the population based on environmental exposures. However, important knowledge gaps regarding the timing for exposures and resultant changes in brain health remain, impeding efforts to develop life-course approaches to maximize AD/ADRD prevention. Acknowledging the NIH-supported research efforts already underway (NIA, 2023a; NINDS, 2024a), further investment is needed to identify and understand brain-critical exposures across the life course (from the prenatal period to late life); vulnerable age windows (Liu et al., 2023a), such as periods of neurodevelopment and major hormonal changes (e.g., adolescence, pregnancy, menopause); the dose-over-time dependency of effects; and how exposures correlate and interact (Tamiz et al., 2022). Cohort studies (discussed in Chapter 2) and natural experiments (e.g., city-level removal of lead pipes, policies aimed at air pollution reduction) provide opportunities to address these scientific priorities, but this requires multidisciplinary research (breaking down discipline-specific silos as discussed in Chapter 5) and attention to addressing current data gaps (e.g., early-life exposures, reproductive history, social determinants of health).
Conclusion 3-1: Understanding differences in pathways that contribute to AD/ADRD across diverse populations requires the expansion of multidimensional diverse cohorts, including cohorts focused on understudied and/or disproportionately affected populations, paired with an investment in tools to generate and link the relevant life-course and exposome information. Also required are the data infrastructure and specialized expertise necessary to carry out complex data integration and analyses.
UNDERSTANDING THE BIOLOGICAL BASIS OF AD/ADRD
Risk and protective factors for AD/ADRD, from individual genes to macro-scale socioeconomic contexts, ultimately affect individuals through their effects on molecules, cells, and physiologic systems (e.g., neural circuits, multiorgan interactions). Molecular targets for pathogenesis (or resilience) include DNA (through mutation or other changes that affect gene expression), RNA, proteins (e.g., misfolding and aggregation, loss of enzymatic function), lipids, and metabolites. Molecular-level changes can affect cells in myriad ways, for example, inducing cell cycle arrest (senescence), cell death, or transition to an altered phenotype. At the organ level, neuronal cell loss and other cellular changes disrupt synaptic function and brain circuitry, as well as the systems that connect the brain with other organs of the body (e.g., vascular, neuromuscular), giving rise to the symptoms experienced by people living with AD/ADRD, including impaired cognitive and motor function and changes in personality and behavior. Despite considerable investment in basic research and resulting scientific advances, there is still much that is not understood regarding these multiscale effects. Continued investment in mechanistic research is needed to understand the biological basis for the effects of risk and protective factors on brain health and to determine whether associations are causal in nature (Tamiz et al., 2022). This needs to include elucidating the biological causes of the different forms of dementia, considering age-associated alterations and the interplay with an individual’s risk factor profile.
The Genetic Architecture of AD/ADRD
While sporadic forms of AD/ADRD are predominant, some forms of dementia are inherited in an autosomal dominant fashion, as is the case for forms of early-onset AD that are mediated by mutations in the APP, PSEN1, and PSEN2 genes (Cacace et al., 2016). As indicated in Table 3-1, there are also autosomal dominant forms of Lewy body dementia (LBD) and frontotemporal dementia (FTD) (Guerreiro et al., 2020). While the occurrence of autosomal dominant mutations is rare, such mutations have
large effect sizes and can therefore be identified and investigated through family studies. Rare population cohorts (e.g., ARTFL-LEFFTDS Longitudinal Frontotemporal Lobar Degeneration [ALLFTD]3, the Dominantly Inherited Alzheimer’s Network) have been established for the study of these dominantly inherited forms of dementia and have yielded invaluable insights on disease mechanisms, biomarkers, and potential treatment targets (Asken et al., 2023; Bateman et al., 2011; Zhang et al., 2023b).
Genetically determined forms of dementia are not limited to those that are inherited in an autosomal dominant manner. Down syndrome-related AD is another form of genetically determined early onset AD that results from a gene dose effect stemming from an extra copy of the APP gene, which is located on chromosome 21. Neuropathological hallmarks of AD (amyloid plaques and tau neurofibrillary tangles) are nearly universal in people with Down syndrome by the age of 40, and the lifetime risk of dementia in this population is estimated at greater than 90 percent (Fortea et al., 2021). As with autosomal dominant forms of AD/ADRD, the natural history of Down syndrome-related AD follows a predictable course (clinical and biomarker changes). Consortia such as the Alzheimer Biomarkers Consortium—Down Syndrome (NIA, 2024a) and the Alzheimer’s Clinical Trials Consortium—Down Syndrome (ACTC-DS, 2024) are working to advance the development of AD biomarkers and therapies for people with Down syndrome.
A recently published analysis proposes that APOE4 homozygotes may constitute a third form of genetically determined AD (Fortea et al., 2024). The E4 allele of the apolipoprotein E gene (APOE) has long been recognized as a major genetic risk factor for AD with a large effect size (Cacace et al., 2016; Lambert et al., 2023). APOE protein is known to be involved in lipid transport (Raulin et al., 2022), but the mechanisms by which APOE4 increase dementia risk have yet to be fully elucidated and represent an area of intense research.4 The proposal of APOE4 homozygotes as a genetic form of AD is based on the near full penetrance of AD neuropathology, as well as the predictability of symptom onset and clinical and biomarker changes over time, characteristics that are also seen with autosomal dominant AD and Down syndrome-related AD. Furthermore, APOE3 and APOE4 heterozygotes exhibit intermediate phenotypes, suggesting the possibility of autosomal semidominant inheritance (Fortea et al., 2024; Genin et al., 2011).
___________________
3 ALLFTD recruits people with autosomal dominant FTD syndromes, as well as people with sporadic FTD.
4 NIA organized a workshop on APOE genetics as a major determinant of AD pathobiology in September 2024. More information is available at https://www.nia.nih.gov/research/dab-dgcg-dn/workshops/apoe-genetics-major-determinant-alzheimers-disease-pathobiology (accessed October 11, 2024).
| Dementia Type | Loci/Genes Associated with Lifetime Risk | ||||
|---|---|---|---|---|---|
| Familial Form | Sporadic Form/by Genetic Ancestry Groups (GWAS Reaching Gnome-Wide Significance) | ||||
| European | African | Hispanic | Asian | ||
| AD | APP, PSEN1, PSEN2 (Cacace et al., 2016) | SORT1, CR1, ADAM17, PRKD3, NCK2, BIN1, WDR12, INPP5D, MME, IDUA, CLNK, RHOH, ANKH, COX7C, TNIP1, RASGEF1C, HLA-DQA1, UNC5CL, TREM2, TREML2, CD2AP, HS3ST5, UMAD1, ICA1, TMEM106B, JAZF1, EPDR1, SEC61G, SPDYE3, EPHA1, CTSB, PTK2B, CLU, SHARPIN, ABCA1, USP6NL, ANK3, TSPAN14, BLNK, PLEKHA1, SPI1, MS4A4A, EED, SORL1, TPCN1, FERMT2, SLC24A4, SPPL2A, MINDY2, APH1B, SNX1, CTSH, DOC2A, BCKDK, IL34, MAF, PLCG2, FOXF1, PRDM7, WDR81, SCIMP, MYO15A, GRN, WNT3, ABI3, TSPOAP1, ACE, ABCA7, KLF16, APOE, SIGLEC11, LILRB2, RBCK1, CASS4, SLC2A4RG, APP, ADAMTS1 (Bellenguez et al., 2022) | ARRDC4/IGF1R, APOE, ABCA7, HMHA1, COBL, SLC10A2 (Mez et al., 2017; Reitz et al., 2013, 2023) | APOE (Reitz et al., 2023) | APOE (East Asians) FAM47E (Japanese), OR2B2 (Japanese+European) RHOBTB3/GLRX, CTC-278L1.1, CTD-2506J14.1, NECTIN2, CHODL, (Chinese) CACNA1A, LRIG1 (Korean) SORL1 (Japanese+Korean+ European) (Jia et al., 2021; Miyashita et al., 2013, 2023; Shigemizu et al., 2021) |
| LBDa | SNCA, GBA, APOE (Guerreiro et al., 2020) | GBA, BIN1, TMEM175, SNCA-AS1, APOE (Chia et al., 2021) | None | None | None |
| FTD | GRN, MAPT, C9ORF72, TBK1, TARDBP, SQSTM1, CHMP2B, VCP, CHCHD10 (Guerreiro et al., 2020) |
MAPT, MOBP, APOE (Manzoni et al., 2024) HLA-DRA/HLA-DRB5, BTNL2 (Ferrari et al., 2014) TMEM106B (FTD with TDP-43 inclusions) (Van Deerlin et al., 2010) |
None | None | None |
NOTES: This table provides a portrait of the genetic architecture of Alzheimer’s disease (AD), frontotemporal dementia (FTD), and Lewy body dementia (LBD)—which includes dementia with Lewy bodies (DLB) and Parkinson’s disease dementia (PDD)—across multiple populations. The table was generated by selecting recent reviews or representative genome-wide association study (GWAS) papers with very large sample size (excluding preprints). For familial forms of AD, LBD, and FTD, the table lists established loci with observed genetic mutations that were found to be significant in family or sequencing studies. For sporadic forms of AD, LBD, and FTD, only loci reaching genome-wide significance (P ≤ 5×10−8) either using samples from one ancestry or after adding samples from multiple ancestries are listed. This is not a comprehensive or systematic review of the literature on risk loci, although it captures the current state of the field and the relative strengths in cohort size and statistical power across populations and diseases.
a Given that DLB and PDD have similar clinical features, Chia and colleagues (2021) examined both disorders together to increase sample size. The two disorders may have different molecular pathways and require different therapeutic strategies, and a better differential diagnostic strategy is needed (Jellinger et al., 2018).
SOURCES: Cacace et al., 2016; Bellenguez et al., 2022; Chia et al., 2021; Ferrari et al., 2014; Guerreiro et al., 2020; Jia et al., 2021; Manzoni et al., 2024; Mez, et al., 2017; Miyashita et al., 2013, 2023; Reitz et al., 2013, 2023; Shigemizu et al., 2021; Van Deerlin et al., 2010.
While requiring further evaluation, the confirmation of APOE4 homozygotes as a form of genetically determined AD would have important implications for approaches to developing and evaluating therapies (e.g., trial participant screening and genetic counseling, population stratification and precision medicine approaches, analytic approaches used in clinical trials) (Fortea et al., 2024). It also underscores the need for continued research to deepen understanding of APOE biology and effects on neurodegeneration (e.g., the contribution of APOE to disease heterogeneity), as well as differences in effects by sex and ancestry. Such research needs to include longitudinal studies of individuals with different APOE genotypes, including not just APOE4 homozygotes and heterozygotes, but also those with alleles (e.g., APOE2) and mutations (e.g., Christchurch mutation in APOE3)5 observed to be protective against clinical dementia (Arboleda-Velasquez et al., 2019; Raulin et al., 2022). Importantly, the study of APOE4 carriers who demonstrate resilience to clinical progression can be a means of identifying protective variants in genes other than APOE (Bhattarai et al., 2024; Huq et al., 2019). Understanding how these rare variants promote dementia resilience opens new avenues for developing treatments. Ensuring adequate inclusion of underrepresented groups in such studies will be important to address pressing scientific questions related to why effect sizes (particularly for APOE4) have been observed to vary across populations with different genetic ancestry. As APOE4 is known to increase risk for related dementias (e.g., LBD) in addition to AD, continued efforts are needed to elucidate the role of APOE in amyloid-dependent and amyloid-independent disease pathways and implications for multiple etiology dementias (Raulin et al., 2022).
Even among those without autosomal dominant mutations or APOE4 alleles, genetic predisposition is thought to be a predominant contributing factor for sporadic (typically late onset) forms of AD/ADRD (Patten and Lein, 2019; Reitz et al., 2023). Genome-wide association studies (GWAS) have identified more than 70 genes or loci associated with sporadic AD (Lambert et al., 2023; Reitz et al., 2023). In many cases, variants identified by GWAS may only have small individual effects on risk but cumulatively—and depending on aging—can contribute to changes in the functions of genes organized in specific molecular and cellular pathways, leading to pathogenic effects (Gan et al., 2018). The large number and diversity of risk genes and loci provide support for the role of multiple mechanistic pathways that may function independently and/or interact with each other or environmental risk factors to contribute to dementia, as discussed later in this chapter.
___________________
5 The observation of a protective effect for the Christchurch mutation in APOE3 for an individual with an autosomal dominant PSEN1 mutation highlights the importance of APOE as a fundamental driver of AD neuropathology.
Given the large number of publications reporting risk genes and loci, efforts are underway to review published studies and evaluate the quality of the evidence for each reported locus or gene (ADSP, 2023). The gene verification committee of the Alzheimer’s Disease Sequencing Project (see Box 3-1) is organizing loci and genes into tiers based on the quality of the evidence of an association. Though results are not yet published in the peer-reviewed literature, this effort will provide the scientific community with lists of loci supported by high-quality evidence that can be used to guide follow-on functional genomics efforts and increase confidence in the pursuit of potential therapeutic targets. The framework developed by the gene verification committee may also serve as a useful model for disease research outside of AD/ADRD.
Identified genetic variants have been used to generate polygenic risk scores that can be used to predict the individual risk of AD (Harrison et al., 2020) and may be helpful tools in the development of precision medicine approaches to prevention and treatment by informing stratification and intervention strategies (discussed further in Chapter 4). However, the majority of known variants were identified in non-Hispanic White individuals from North America and Europe (see Table 3-1), suggesting that additional genetic risk factors for other populations remain to be discovered and limiting the applicability of current polygenic risk scores to individuals of other ethnicities (Clark et al., 2022; Reitz et al., 2023). Additionally, there may be sex differences in genetic risk factors that would need to be accounted for (Mielke, 2024).
Far fewer risk loci have been identified for sporadic forms of LBD, FTD,6 and vascular dementia—in part because of smaller sample sizes for GWAS as compared to AD. In contrast to risk loci with small effect sizes, strong risk loci, such as those associated with autosomal dominant forms, can be identified without the need for large sample sizes. As indicated in Table 3-1, there is some evidence of overlap in risk genes for these different forms of dementia (Chia et al., 2021; Ciani et al., 2019; Guo et al., 2022; Ikram et al., 2017; Rongve et al., 2019), but more studies are needed to firm up these relationships. Additionally, genes associated with common diseases, such as cardiovascular disease and stroke, are shared with AD/ADRD. Thus, there is a critical need to conduct further GWAS of adequate sample size and statistical power to detect additional genetic associations for sporadic forms of AD/ADRD (Bellenguez et al., 2022). It will be important in such efforts to use large study populations (to detect rare variants) representing
___________________
6 Sporadic forms make up 70 percent of FTD, a smaller fraction as compared to AD and LBD (Greaves and Rohrer, 2019). A larger number of strong risk loci (i.e., those associated with autosomal dominant forms of disease) have been identified for FTD as compared to AD (see Table 3-1).
BOX 3-1
Alzheimer’s Disease Sequencing Project
The NIA-funded Alzheimer’s Disease Sequencing Project (ADSP) was established in 2012 following the enactment of the National Alzheimer’s Project Act and has grown into a global collaboration focused on better understanding the genetic architecture of AD/ADRD (ADSP, 2024). The ADSP works to identify and explore new genes and genetic variations that are linked to increased risk of or protection against AD/ADRD with the goal of advancing findings into therapeutic targets for further development. The ADSP network of funded collaborations and programs includes infrastructure and expertise to genotype and sequence samples from existing studies from around the world; perform functional and computational analyses; and process, store, and share these high-quality data with researchers (ADSP, 2024). Since its initiation in 2012, ADSP has evolved over a series of phases. The first two phases included a majority of samples from participants of non-Hispanic White ancestry. ADSP received additional funding from NIA to diversify the existing ADSP dataset (NIA, 2024b). This most recent phase, ADSP Follow-Up Study 2.0: The Diverse Population Initiative, involves the whole-genome sequencing of 18,500 AD/ADRD cases and 18,500 controls each from Hispanics, African Americans and Africans, and Asians using samples from the U.S. and international collaborations in Africa, Central and South America, and Asia (NIA, 2024b). This expansion in diversity will enable the identification of genetically driven targets and improvements in the design of clinical trials (ADSP, 2024; NIA, 2024b). The ADSP Follow-Up Study 2.0 “follows on three other recent initiatives to enhance the ADSP’s capability to identify new risk and protective genes:
- The Phenotype Harmonization Consortium is aggregating and harmonizing clinical, cognitive, imaging, and biomarker phenotype data from all participating ADSP cohorts.
- The Functional Genomics Consortium is generating “omics” experimental data to further characterize ADSP genetic findings and to help better define subtypes of Alzheimer’s and related dementias.
- The Artificial Intelligence/Machine-Learning Consortium employs sophisticated computational methods to further integrate and analyze ADSP data and optimize subject selection for clinical trials based on participants’ characteristics” (NIA, 2024b).
diverse ancestral backgrounds, as genetic analysis has shown that continental genetic ancestry plays an important role in AD risk and protection, as evidenced by the variation in risk effect for the APOE4 allele across populations with diverse ancestral backgrounds (Reitz et al., 2023). Observing more population-specific variants increases statistical power in AD/ADRD gene discovery. The Alzheimer’s Disease Sequencing Project has been a major NIA investment in improving understanding of the genetic architecture of AD/ADRD, and recent phases of the initiative have included a focus on expanding the genetic diversity of study populations (see Box 3-1).
Continued investment in understanding how different variants affect the functional role of each gene associated with disease risk can help to elucidate the biological mechanisms of AD/ADRD (discussed later in this chapter) and thereby guide the development of more targeted intervention strategies. For example, genetic studies have implicated lysosomal dysfunction and autophagy pathways in AD/ADRD (Bellenguez et al., 2022; Deng et al., 2017). However, caution is required when attempting pathway analysis using GWAS findings as it is not always clear which gene is linked to an identified locus and pathway annotation suffers from a number of limitations (Silberstein et al., 2021). Further development of tools and processes is needed to improve the translation of genetic signals to target pathways. In the meantime, triangulation using a combination of genetic findings and data from experimental studies (in vitro and in vivo) can increase confidence in the identification of affected pathways.
Understanding Gene–Environment Interactions
As noted earlier, there are various molecular and cellular targets through which exposome factors (e.g., lifestyle, social determinants of health, pollutant exposures) may exert pathogenic effects. For example, exposome factors can interact directly with proteins (e.g., bind to and/or alter the function of receptors, enzymes, and structural proteins), but may also alter protein levels or function through interactions with an individual’s epigenome and/or genome. In framing brain health as a trajectory over the life course, an individual’s genetics can be envisioned as defining the major bounds for that trajectory, while the exact course will be determined by the interactions between genes and exposome factors. Perhaps the best studied gene–environment interactions are those involving the APOE gene, but generally these interactions remain poorly understood (Dunn et al., 2019; Migliore and Coppedè, 2022). In fact, environmental and social factors (e.g., socioeconomic status) likely influence observed associations between genetic variants and AD (Reitz et al., 2023), making it challenging to disentangle risks from different sources, which is important for understanding the variation in AD/ADRD risk across diverse populations.
Although population-specific gene variations may have different levels of impact in disturbing disease pathways, another possibility is that population-specific exposures and gene–environment interactions contribute to observed population-specific differences. Deciphering the individual causes and trajectories of AD/ADRD is dependent on understanding an individual’s specific profile of risk genes and their exposures (e.g., family resources, education, chemical exposures, inflammation, trauma), including comorbid conditions (e.g., metabolic disorders), across the life course.
The field of functional genomics is advancing understanding of the mechanisms by which the exposome interacts with an individual’s genetic background (Reitz et al., 2023). Such knowledge will be critical to uncovering how gene–environment interactions contribute to the development of AD/ADRD. Quantification of messenger RNA (mRNA) and protein levels through transcriptomic and proteomic approaches, respectively, can provide information on gene expression levels that can be linked to specific exposures. Epigenetics methods can be used to directly examine allele-specific DNA methylation and histone acetylation, which modulate gene expression (Reitz et al., 2023). A growing body of evidence suggests gene–environment interactions commonly manifest through epigenetic mechanisms (a description of how epigenetic mechanisms influence gene expression and pathogenesis can be found in Box 3-2) (Cavalli and Heard, 2019; Maloney and Lahiri, 2016; Marsit, 2015; Migliore and Coppedè, 2022; Schrott et al., 2022). Air pollution exposure, for example, has been linked to the DNA methylation of genes involved in inflammation pathways, contributing to the risk of cerebrocardiovascular disease (Migliore and Coppedè, 2022; Vineis et al., 2020).
Early-life lead exposure has also been associated in animal models with decreased methylation at the promoter of APP and histone modifications that influence the expression of genes related to the AD pathway (APP and BACE1) (Mei et al., 2023). These kinds of epigenetic effects are not limited to exposures involving environmental contaminants. Lower folate and vitamin B12 levels, for example, have been linked to reduced methylation of genes involved in the production of amyloid (PSEN1 and BACE1) in AD patients as compared to age-matched controls (Migliore and Coppedè, 2022). Proteomics can enable the identification of aberrant posttranslational modifications (e.g., phosphorylation, glycosylation, acetylation, and ubiquitination)—mechanisms that regulate the trafficking, function, and degradation of proteins in normal cellular processes—which may result from exposures and influence pathological pathways leading to AD/ADRD (Guan and Wang, 2023; Ramesh et al., 2019). Thus, the layering of multiomics data has great potential to elucidate gene–environment interactions in AD/ADRD. However, limited access to datasets that include both genetic and exposome data is a current barrier to these kinds of
BOX 3-2
Epigenetic Influences and Gene–Environment Interactions
The epigenome encompasses all of the chemical modifications that are added to the genome and the changes to histone proteins that influence chromatin structure. Epigenetic processes enable cells to incorporate external stimuli into their genetic material, thereby influencing gene expression without modifying the DNA sequence itself (Maloney and Lahiri, 2016; Rozek et al., 2014). These dynamic modifications are reversible in nature and encompass DNA methylation, chromatin remodeling, histone modification, and noncoding RNA, such as microRNAs. Changes to chromatin structure influence the accessibility of DNA by the cell’s transcription machinery, thereby modulating gene expression. Similarly, DNA methylation—the reversible addition of a methyl group to the cytosine base of a cytosine-guanine pair, which are common in regulatory regions of genes (Rozek et al., 2014)—influences the binding of transcription factors that mediate the initiation of the transcription process. Transcription (and gene expression) may be up- or down-regulated depending on whether transcription factor binding affinity is increased or decreased by the methylated cytosine, but more commonly DNA methylation reduces gene expression (Maloney and Lahiri, 2016; Rozek et al., 2014).
Noncoding microRNAs can regulate gene expression through binding to mRNA but also play a role in epigenetic regulation processes through posttranscriptional regulation of important chromatin- and DNA-modifying enzymes responsible for the epigenetic modification of DNA. These miRNAs are themselves regulated through epigenetic mechanisms (methylation) (Favier et al., 2021; Migliore and Coppedè, 2022). Through these mechanisms, cells can respond and adapt to diverse environmental cues, which play a vital role in gene regulation and cellular function (Bufill et al., 2020). However, epigenetic mechanisms can contribute to pathogenesis by altering gene expression and the levels of the encoded protein.
It is possible that epigenetic changes induced in early life (e.g., from stress or environmental exposures) can generate susceptibility to other insults that accumulate over the life course, eventually leading to disease, consistent with the developmental origins of health and disease theory discussed earlier in this chapter (Migliore and Coppedè, 2022). Postmortem studies have identified altered epigenetic profiles in brain tissue from individuals diagnosed with AD and other forms of dementia premortem, and there is experimental evidence implicating epigenetic changes in the pathogenesis of AD and related dementias (Gao et al., 2022; Martinez-Feduchi et al., 2024; Nikolac Perkovic et al., 2021).
The dynamic nature of the epigenome over the human lifespan and the tissue-specificity of epigenetic changes raises unique challenges related to standardization of epigenetic measurement methods and data analysis (Carter et al., 2017). Artificial intelligence and machine learning methods that are enabling integrated analysis of epigenetic and other omics data have the potential to overcome some data analytic challenges and advance precision medicine approaches (Hamamoto et al., 2020).
analyses (Pericak-Vance, 2024). Moreover, collaboration among biomedical science experts and experts in bioinformatics and information science will be needed to tackle the data analytic challenges related to managing and integrating these vast and complex datasets and developing tools to extract insights (Hamamoto et al., 2020).
In addition to research focused on understanding how interactions between exposures and genetics lead to disease, other studies are exploring the degree to which genetic risk can be offset by protective aspects of the exposome and the mechanisms behind those protective effects. For example, a recent observational study of associations by Lourida and colleagues (2019) found that a healthy lifestyle (e.g., regular exercise, healthy diet, no current smoking, and alcohol consumption in moderation) could lower but not eliminate dementia risk in individuals with a high genetic risk.
Understanding Molecular and Cellular Mechanisms Associated with Aging and AD/ADRD
Neurodegenerative diseases such as AD/ADRD exhibit a wide range of clinical manifestations, which arise from the loss of specific neurons and synapses in different areas of the brain. While different disorders display some tissue specificity (e.g., degeneration of cholinergic neurons for AD, degeneration of dopaminergic neurons for Parkinson’s disease [PD], degeneration of motor neurons for amyotrophic lateral sclerosis [ALS]), they also feature some anatomical overlaps of affected brain regions and possess shared characteristics and mechanisms, particularly the regional aggregation and spreading of cytosolic or nuclear inclusion proteins. Thus, even when different diseases are associated with distinct genetic variants, common biological themes can be observed (Behl, 2023; Gan et al., 2018).
As discussed in Chapter 1, mixed pathologies are now believed to be predominant; based on the current biological definition of AD as plaques- and-tangles disease, “pure Alzheimer’s is rare” (Robinson et al., 2021). There is a strong pathological overlap of AD with, for instance LBD, vascular dementia, or hippocampal sclerosis (Jellinger, 2022; Rabinovici et al., 2017). Similarly, Lewy bodies comprising alpha-synuclein protein are features of LBD and Parkinson’s disease dementia (Mensikova et al., 2022). Given the prevalence of mixed pathologies in AD/ADRD and the potential for variation in aberrant protein aggregates found in different diseases, it may be possible to achieve broader effect through interventions targeting underlying biological pathways not specific to a single pathology. Such an approach may benefit from moving away from historical clinical classifications and definitions toward descriptions that reflect diseases at a molecular and cellular resolution (Balusu et al., 2023).
Genetic mutations and epigenetic alterations can lead to defects in a multiplicity of molecular and cellular pathways, depicted in Figure 3-3, that are common across neurodegenerative diseases and occur in an age-dependent manner (Behl, 2023; Gan et al., 2018; Schumacher et al., 2021). These shared molecular and cellular pathways include:
- chronic inflammation and immune dysfunction,
- vascular dysfunction and diminished blood–brain barrier integrity,
- aberrant proteostasis and deficiency in the endosomal–lysosomal (autophagy) pathway,
- mitochondrial and metabolic dysfunction,
- disturbed lipostasis and altered lipid metabolism,
- cellular senescence, and
- epigenetic dysregulation (Hara et al., 2019).
There is increasing evidence that each of these potentially pathogenic factors and processes can play a role in the development of AD/ADRD and exert effects in varying combinations over the life course. Currently lacking, however, is an understanding of the interactions and sequential cascading effects that translate dysregulation at any one level into disease. A better understanding and an integrated model (discussed later in this chapter) will be needed to guide intervention strategies that reflect the multifactorial nature of disease. As new therapeutics targeting these earlier-stage molecular and cellular pathways are developed, it will be important to have biological markers that could be used in trials to show that the intended pathway is engaged and the therapy is having the expected effect (Lamb, 2024).

NOTE: This is a network of interacting cells; a number of interacting factors and pathways affect neuronal function and, eventually, lead to dysfunction and degeneration. The pathogenic processes occur over time and can change during aging. BBB = blood–brain barrier; EL = endosome-lysosome; TDP43 = TAR DNA-binding protein 43.
SOURCE: Image reproduced with permission from Christian Behl.
Chronic Inflammation and Immune Dysfunction
Multiple studies have established links between the presence of proinflammatory mediators in the bloodstream and the advancement of neurodegenerative disorders, indicating that systemic inflammation may contribute to the emergence of persistent brain inflammation (Cao and Zheng, 2018; Italiani et al., 2018; Kim et al., 2018; Swardfager et al., 2010). Chronic inflammation, which plays a role in various age-related ailments,
has been associated with decreased brain volume and impaired cognitive function in AD/ADRD (Franceschi and Campisi, 2014; Hara et al., 2019; Pilling et al., 2015).
As the resident immune cells in the brain, microglia play essential roles in maintaining homeostasis, including defense against infection or damage and phagocytosis of debris or dying cells (Salter and Stevens, 2017). Microglia also secrete tissue rebuilding factors (Vasic et al., 2019), a function that may be targeted by therapies aimed at stimulating brain tissue regeneration (discussed further in Chapter 4). During aging and in the context of some neurodegenerative diseases, microglia can acquire an activated proinflammatory phenotype characterized by morphological and functional changes, which include the release of proinflammatory cytokines and other neurotoxic mediators (Tejera et al., 2019; Wang et al., 2023). While pathways leading to microglial activation are not fully understood, there is some evidence that systemic inflammation can affect the inflammatory response of microglial cells in the brain and interfere with their clearance of aggregating proteins, such as amyloid beta in AD (Tejera et al., 2019). Environmental exposures such as brain chemical injuries or infection that trigger systemic inflammatory responses may thereby contribute to microglial activation (Ahmed et al., 2024; Greve et al., 2023; Jayaraj et al., 2017; Zhang et al., 2023c). Similarly, studies have found a link between proinflammatory microbial metabolites in the gastrointestinal tract and microglial activation and alpha-synuclein aggregation in the brain in PD (Sampson et al., 2016). Perivascular macrophages and astrocytes are also involved in the brain’s innate immune response. Depending on the injury, astrocytes can demonstrate distinct activation states and transcriptional signatures that mediate neurotoxic or neurotrophic effects, which may be modulated by a microglia-astrocyte crosstalk mechanism (Gao et al., 2023; Liddelow et al., 2017).
While not as well studied as the role of the innate immune system in AD/ADRD, the adaptive immune system may also contribute to neurodegenerative disorders and may have both protective and pathogenic roles (Chen and Holtzman, 2022; Mayne et al., 2020). Studies of the adaptive immune system in dementia have primarily focused on the composition and roles of T-cell populations, which appear to infiltrate the brain from the periphery, potentially facilitated by changes to the blood–brain barrier (Chen and Holtzman, 2022). Activated T cells may perpetuate the inflammatory cascade and induce microglia into an activated phenotype by secreting proinflammatory and neurotoxic mediators. Further research is needed to understand the function of specific T-cell populations in different types of dementia. For example, proinflammatory CD4+ TH17 cells have been implicated in neuronal degeneration in LBD (Gate et al., 2021). CD4+ T cells that exhibit an immune suppressive phenotype, known as Tregs,
are thought to have a neuroprotective role in AD based on animal studies. However, alterations in Treg levels and immunomodulatory activity over the course of neurodegeneration are not well understood and some studies have suggested a detrimental effect of Tregs (Duffy et al., 2018; Gendelman and Appel, 2011; Mayne et al., 2020).
The role of B cells and humoral immune responses in neurodegeneration leading to dementia is less studied (Chen and Holtzman, 2022; Lutshumba et al., 2021), but research in mouse models suggests a role for B cells in AD progression (Kim et al., 2021). Similarly not well understood is the potential role of autoimmunity in neurodegenerative diseases that cause dementia. Some converging lines of evidence suggest that dementia may have an inflammatory autoimmune component (Lindbohm et al., 2022). Links between autoimmune disorders and subsequent dementia based on epidemiological studies, however, have not been as strong as the associations between infections and dementia (Janbek et al., 2023).
Genetic studies provide strong evidence for the link between immune dysfunction, inflammation, and AD/ADRD. Indeed, many genes associated with neurodegenerative diseases regulate responses of the innate immune system (Gan et al., 2018). For example, whole-genome sequencing studies helped identify rare variants in TREM2 (a transmembrane receptor highly expressed in immune system cells, including microglia and myeloid cells) that lead to a two- to threefold increase in AD risk (Gan et al., 2018; Guerreiro et al., 2013; Jonsson et al., 2013; Zhang et al., 2013). In familial FTD-TDP, genetic mutations that lead to insufficient production of the progranulin gene product, which is highly expressed in microglia, are associated with exacerbation of proinflammatory responses and the activation of microglial cells (Baker et al., 2006; Cruts et al., 2006; Yin et al., 2010). The immunomodulatory effects of these and other disease-associated gene mutations implicate aspects of the innate immune system in neurodegenerative diseases (Gan et al., 2018).
To further elucidate the role of inflammation and immune dysregulation in AD/ADRD, key knowledge gaps need to be addressed, including:
- pathways promoting various microglial states in neurodegeneration;
- connections between microglia, astrocytes, neurons, oligodendrocytes, and endothelial cells;
- the role of microglia in the clearance of protein aggregates; and
- the role of the adaptive immune system in neurodegeneration (Lamb, 2024).
Such research may suggest strategies for therapies targeting neuroinflammation (Lin et al., 2023; Tejera et al., 2019). While broad-spectrum anti-inflammatory drugs have not been effective, specifically targeting
some aspects of inflammation may be a more promising approach (Fillit et al., 2023).
Vascular Dysfunction
Vascular pathology is widely acknowledged as a contributing factor to the development of dementia (Gorelick et al., 2011; Hara et al., 2019). Epidemiological evidence supports an association between risk factors for cardiovascular disease, cerebrovascular dysfunction, and cognitive impairment (Corriveau et al., 2016; Pacholko and Iadecola, 2024). Vascular pathologies are thought to be widely prevalent. Findings from two longitudinal cohort studies showed that over 95 percent of individuals diagnosed with probable AD had mixed pathologies observed at autopsy. Approximately 90 percent of those with probable AD had vascular pathologies present (Kapasi et al., 2017).
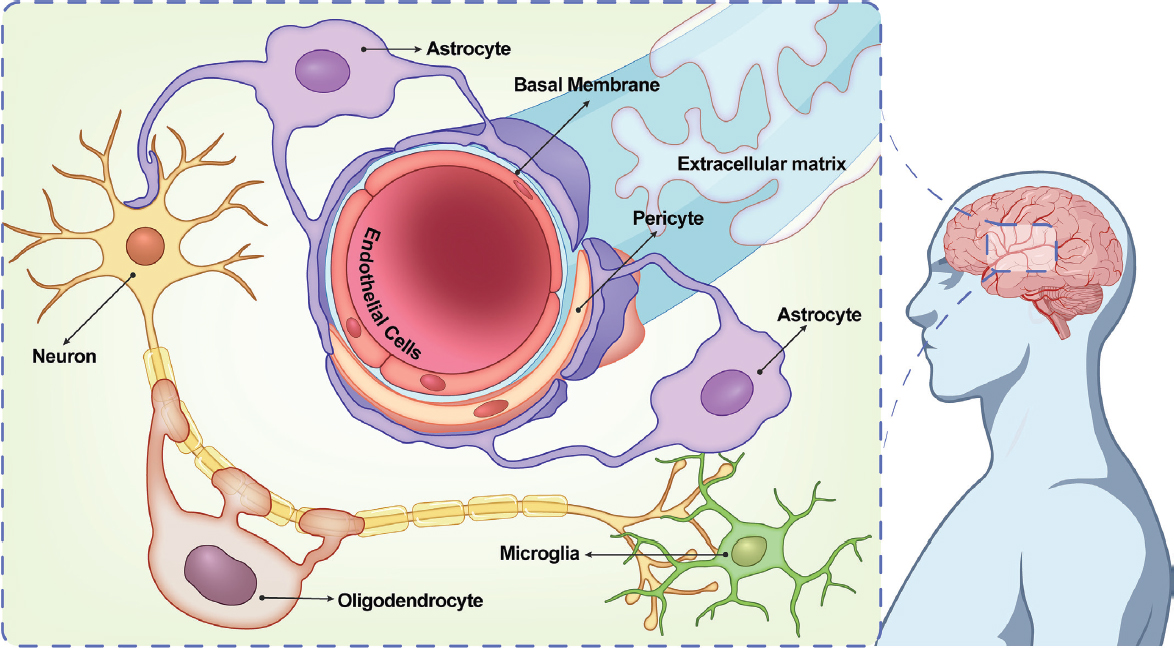
Vascular contributions to cognitive impairment and dementia arise from age-related alterations in the neurovascular unit. The neurovascular unit consists of various components, including nonfenestrated endothelial cells,7 pericytes, smooth-muscle cells, astrocytes, microglia, oligodendroglia, and neurons (see Figure 3-4). These components collectively contribute to the regulation of cerebral blood flow matched to neuronal metabolic demands (Claassen et al., 2021), maintenance of the blood–brain barrier, and communication between the vascular and neural compartments. Age-related changes within the neurovascular unit can disrupt these crucial functions, leading to cognitive impairment and the development of vascular dementia (Cai et al., 2017). During aging, and particularly in the context of neurodegenerative diseases, there is a notable decline in the population of pericytes, resulting in diminished cerebral blood flow delivery and subsequent downstream changes leading to neurodegeneration and dementia (Uemura et al., 2020).
One vascular pathology relevant to AD is vascular breakdown in the brain resulting from blood–brain barrier disruption (Fillit et al., 2023), which may be caused by pathological changes to the neurovascular unit. The integrity of the blood–brain barrier diminishes during normal aging, and this decline becomes even more pronounced in individuals with AD and related dementias, which often results in capillary leakage, brain leukocyte infiltration (Cai et al., 2017), ingress of toxic substances, and reactive response of astrocytes and microglia, which further aggravates neural dysfunction (Watanabe et al., 2020). Additionally, such breakdown also leads to the leakage of fibrinogen into the central nervous system (CNS),
___________________
7 “Non-fenestrated endothelium is characterized by low permeability and a high abundance of tight junctions” (Gifre-Renom et al., 2022).
triggering neuroinflammation and causing insoluble fibrin clots to form (Bardehle et al., 2015; Fillit et al., 2023). Fibrinogen is not detectable in a healthy brain; in AD, however, it reaches detectable levels and interacts with amyloid, which then exacerbates clotting, fibrin deposition, and proinflammatory signaling (Fillit et al., 2023; Ryu and McLarnon, 2009; van Oijen et al., 2005; Viggars et al., 2011).
Decreased blood flow to the brain and the disruption of the blood–brain barrier can on their own contribute to neurodegeneration and the development of dementia, but may also be elements of what has recently been described as “multiple hits” models (which include vascular changes) contributing to dementia (Patrick et al., 2019; Steele et al., 2022). Multihit models (see Figure 3-5) describe the convergence of interconnected hits associated with age-related, genetic, vascular, and immune factors (e.g.,

SOURCE: Mehta and Mehta, 2023. CC BY 4.0.
neurovascular dysfunction, neuroinflammation, oxidative stress, endosomal trafficking dysfunction, changes in lipid metabolism, among others) to precipitate AD pathology, including extracellular amyloid plaques and intraneuronal neurofibrillary tangle formation, as well as other alterations that contribute to dementia (Steele et al., 2022).
One prominent NIH-supported effort in this area is the Molecular Mechanisms of the Vascular Etiology of Alzheimer’s Disease (M2OVE-AD) initiative, which was established in 2016 and works to examine the link between vascular risk factors, cerebrovascular disease, and AD (NIA, 2016). This consortium brought together over 12 crossdisciplinary research teams from different institutes to use brain tissue and peripheral fluids collected from previous studies that have characterized neurodegeneration and vascular disease to generate various types of multiomics data. Integration of these data with cognitive, neuroimaging, and vascular physiology data is helping to better understand and predict the underlying molecular mechanisms behind vascular pathology and AD risk (AD Knowledge Portal, 2024a).
Aberrant Proteostasis and Deficiency in the Endosomal–Lysosomal (Autophagy) Pathway
The accumulation of misfolded proteins in the CNS is a common feature of many neurodegenerative diseases, implicating a major role for a breakdown in cellular protein quality control and clearance pathways (Gan et al., 2018; Morawe et al., 2012). Protein folding is mediated by molecular chaperones, which are themselves proteins that can become dysregulated. One therapeutic approach under investigation is to use the activity of molecular chaperones to disaggregate misfolded proteins and refold them into their native forms. An alternative approach involves targeting the cellular machinery involved in clearing protein aggregates. Autophagy is the cellular mechanism responsible for breaking down and recycling aggregated misfolded proteins and damaged organelles and plays a major role in proteostasis. In conjunction with the ubiquitin-proteasome and the lysosomal systems, autophagy functions to alleviate cellular stress by breaking down pathogenic forms of aggregate-prone proteins (i.e., TDP-43, amyloid, tau, and alpha-synuclein), lipids, dysfunctional mitochondria, and other organelles in cells (Hu et al., 2015; Scrivo et al., 2018).
This cellular mechanism is particularly critical in postmitotic cells such as neurons, as they are unable to reduce proteotoxic burden and eliminate cellular waste through cell division, rendering them more vulnerable to proteotoxic insults (Nixon, 2013). Thus, functional protein and organelle turnover as provided by a functional autophagy secures neuronal function, stability, and resistance. During the aging process, autophagy may become disrupted, resulting in intracellular buildup of various misfolded
proteins. This age-associated effect can be magnified in the context of neurodegenerative disease (Bartlett et al., 2011; Gonzales et al., 2022).
Aberrant autophagy may contribute to the pathologic accumulation of amyloid plaques, tau tangles, and other protein aggregates found deposited in the brain (e.g., alpha-synuclein, TDP-43) in AD and other forms of neurodegenerative disorders (Fillit et al., 2023; Rubinsztein et al., 2011). Analysis of postmortem human brains affected by AD reveals abnormal autophagy (Lee et al., 2022; Loeffler, 2019). Propagation of misfolded proteins in animal models match observed progression patterns of neurodegeneration in the human brain (Yang et al., 2021). Endosomal–lysosomal dysfunction is one of the early pathophysiological changes that can be observed in neurodegeneration. The lysosome is the executor of autophagic clearance of proteins. Lysosomal dysfunction is acknowledged as a central catalyst of neurodegeneration (Nixon, 2020), and, consequently, lysosomal activity is a new pharmacological target.
Support for this pathway comes from observed mutations or perturbations in autophagy pathway genes in various neurodegenerative diseases. For example, genes implicated in Parkinson’s disease (e.g., PARK2, ATP13A2, PINK1, GBA) encode lysosomal proteins or regulators of endolysosomal trafficking (Abeliovich and Gitler, 2016). In progressive supranuclear palsy, part of the FTD complex of neurodegenerative disorders, GWAS have implicated genes for components of the ubiquitin–proteasome system, including TRIM11 (Jabbari et al., 2018), a gene that also has been shown to be down-regulated in AD (Zhang et al., 2023d). While not fully understood, APOE4 is thought to contribute to dysregulation in the endosome–lysosome and autophagy pathways, which may mediate increased AD risk in APOE4 carriers (Asiamah et al., 2024).
Protein degradation and clearance mechanisms (e.g., stabilization of lysosomal function) are becoming prime pharmacologic therapeutic targets as they have the potential to affect multiple proteinopathies simultaneously (Knopman et al., 2021), which could be especially promising for those with multiple brain pathologies. This is in contrast to approaches such as a focused anti-amyloid or anti-tau immunotherapy that target only one type of protein aggregate.
Mitochondrial and Metabolic Dysfunction
Mitochondrial function is closely intertwined with cell viability, overall cellular function, and the various hallmarks associated with aging. Mitochondria not only play a critical role in cellular respiration—using oxygen to extract, transfer, and generate energy from such molecular substrates as glucose, fat, fatty acids, and amino acids—they are also involved in maintaining calcium and iron balance, regulating cell proliferation and
cell death, facilitating cell signaling, and ensuring proteostasis (Gonzales et al., 2022; Lima et al., 2022). Neurons have a high metabolic demand, and dysfunctional mitochondria disturb the energy supply of neurons. For neurons and glial cells to function optimally, a plentiful and uninterrupted supply of adenosine triphosphate (ATP) is needed, which is best accomplished by oxidative phosphorylation of glucose. As compared to healthy aging, glucose consumption is reduced (i.e., hypometabolism) in AD/ADRD (Kato et al., 2016; Yan et al., 2020). Using fluorodeoxyglucose positron emission topography, disruption to cerebral glucose metabolism in specific brain regions has been detected (Bohnen et al., 2012; Kato et al., 2016; Yan et al., 2020). Reduced glucose utilization can occur prior to the presentation of clinical symptoms as demonstrated in a study involving participants with mild cognitive impairment due to AD (Drzezga et al., 2003; Yan et al., 2020). A longitudinal study assessing data from 128 participants found that glucose uptake in the precuneus, an area of the brain in which there is early amyloid deposition, is reduced in patients 10 years prior to the development of symptoms (Bateman et al., 2012; Yan et al., 2020).
Neurogenic glucose metabolism is perturbed by impaired insulin signaling, resulting in characteristics of AD that mirror the pathophysiology of non-CNS tissues in type 2 diabetes mellitus (Duelli and Kuschinsky, 2001). Moreover, reductions in glucose transporters GLUT1 and GLUT3 were observed in AD patient brains (An et al., 2018) and has been associated with a reduction in brain glucose consumption and cognitive impairment (Landau et al., 2010; Yan et al., 2020). Brain insulin resistance is acknowledged as an early and common feature of AD and can be closely linked to cognitive decline. In fact, there are many pathogenetic connections between brain disorders—including neurodegeneration—diabetes, and insulin resistance (de Galan, 2024).
Oxidation end products are pathological hallmarks of a variety of neurodegenerative pathologies. Mitochondria are a key source of oxidative stress resulting from respiratory chain activities and potential generation of superoxide radicals. Impairment of the activity of mitochondrial respiratory chain complexes are reported in AD, PD, and ALS (Golpich et al., 2017). Sublethal but progressive changes in mitochondrial function have been proposed to drive aging (i.e., the free radical theory of aging) (Harman, 1956) and AD (i.e., the “mitochondrial cascade” hypothesis of AD) (Swerdlow and Khan, 2004). Destabilized mitochondria create more free oxygen radicals than can be buffered sufficiently by the intrinsic antioxidant systems. A previous study reported that impaired mitochondrial function resulting from mitochondrial DNA (mtDNA) damage tends to increase with age, and levels of mtDNA damage were increased in individuals with AD as compared to age-matched controls (de la Monte et al., 2000). Single-cell
analyses have revealed an increase in mtDNA deletions specifically within neurons affected by AD (Krishnan et al., 2012).
In experiments where mtDNA is transferred from donor cells to cells with identical nuclear DNA but lacking mtDNA, it has been demonstrated that mtDNA from individuals with AD is responsible for subtle variations in mitochondrial morphology, biogenesis, membrane potential, oxidative stress, and calcium buffering capacity (Swerdlow et al., 2017). Such distinct mitochondrial characteristics seen in peripheral tissues of individuals with AD, as opposed to control groups, implies that changes in mitochondrial status relevant to the brain can potentially be detected and monitored in peripheral samples. These findings suggest that mitochondrial dysfunction, which may be a cause rather than a result of AD neuropathology, could be an early event in the disease onset and progression (Theurey and Pizzo, 2018; Wilkins and Swerdlow, 2021).
Disturbed Lipostasis and Altered Lipid Metabolism
The brain has an abundance of lipid-rich myelin. Changes in the lipid composition of the brain are observed in both the natural process of aging and various neurodegenerative diseases (Gonzales et al., 2022). Genetic studies—including genetic linkage studies, GWAS, and exome sequencing studies—have repeatedly linked genes/loci related to lipid metabolism (including APOE) with AD (Andrews et al., 2020; Gonzales et al., 2022; Jones et al., 2010; Sims et al., 2020), suggesting a role for aberrations in lipid transport, lipid processing, and metabolism. The field of lipidomics, which involves the investigation of “pathways and networks of cellular lipids in biological systems” (Gonzales et al., 2022), has uncovered AD-associated lipid profiles (Han, 2010; Naudi et al., 2015). For instance, multiple research groups have consistently reported the early accumulation of ceramide levels in brains affected by AD (Cutler et al., 2004; Han et al., 2002). In contrast, sulfatides, a group of sulfoglycolipids highly enriched in myelin, have been shown to be significantly reduced at the earliest clinical stages of AD (Cheng et al., 2013; Gonzales et al., 2022; Han et al., 2003; Irizarry, 2003). Similarly, phospholipid plasmalogen was found to be reduced in the brains of individuals with AD and is associated with disease severity (Goodenowe et al., 2007).
Although the role of lipid metabolism in the pathogenesis of AD/ADRD is still under study, one interesting avenue of lipidomics research is the potential application of lipid mediators to resolve inflammation associated with proteinopathy. Free forms of proresolving lipid mediators made from the enzymatic oxygenation of polyunsaturated fatty acids have been found to be deficient in AD as a result of depletion in the pool of bound or esterified lipid mediators. Improving understanding of the regulation of
lipid esterification and release could suggest effective therapeutic strategies for inflammation resolution (Shen et al., 2022; Taha, 2024).
Cellular Senescence
Cellular senescence can be defined as “a cell state triggered by stressful insults and certain physiological processes, characterized by a prolonged and generally irreversible cell-cycle arrest with secretory features, macromolecular damage, and altered metabolism” (Gorgoulis et al., 2019, p. 814). Senescent cells employ antiapoptotic pathways and halt the cell cycle as a means to evade cell death. Additionally, these cells release various substances such as proinflammatory cytokines, chemokines, growth factors, extracellular remodeling proteins, and other signaling molecules, collectively known as the senescence-associated secretory phenotype (SASP) (Gorgoulis et al., 2019). In the absence of efficient senescent cell clearance, SASP can lead to tissue damage, cell death, or even the conversion of other cells into a senescent state as a propagating phenotype (Yousefzadeh et al., 2020). As people age, senescent cells are found to accumulate throughout the body, including the brain (Yousefzadeh et al., 2020).
Nerve cells are aging over the lifetime and are only stable and functional if the nerve-cell intrinsic defense, repair, and recycling mechanisms are adequately working. Such repair mechanisms include local (axonal, synaptic, soma compartment) protein recycling pathways (see the section above on proteostasis and autophagy) but also DNA surveillance pathways. In postmortem human brain tissues of AD, various senescent cell types have been identified, including astrocytes (Bhat et al., 2012), neurons (Dehkordi et al., 2021; Musi et al., 2018), microglia (Streit and Xue, 2014), oligodendrocyte precursor cells (Zhang et al., 2019), and endothelial cells (Bryant et al., 2020). Unbiased single-cell transcriptomics analysis conducted on the dorsolateral prefrontal cortex of individuals with AD has shown that excitatory neurons are a predominant senescent cell type in the brain and are correlated with the coexistence of intracellular neurofibrillary tangles (Dehkordi et al., 2021). In healthy aging brains, however, analyses of transcriptomics data derived from bulk tissue revealed that the prevailing senescent cell types in the brain were endothelial cells and microglia (Xu et al., 2022). This finding underscores the potential disparities in senescent cell types observed in healthy versus diseased states, but it could also be attributable to the nature of the analyzed material and variations in the predetermined criteria used to define senescence.
While tracing the cause of brain senescent cell emergence in humans is not currently feasible, studies conducted on rodents revealed that such emergence could be triggered by the endogenous accumulation of tau (Bussian et al., 2018; Musi et al., 2018) or amyloid beta protein (Zhang et
al., 2019), insulin resistance (Chow et al., 2019), and immune dysfunction (Yousefzadeh et al., 2021). Contributing exposomal factors may include a high-fat diet (Ogrodnik et al., 2019), chronic ethanol exposure from alcohol consumption (Sun et al., 2023), chronic unpredictable stress (Lin et al., 2021), and brain injury (Arun et al., 2020). Further elucidation of the role of cellular senescence may be achieved through comparative studies, including widescale multiomics studies, of brain regions that are normally spared in AD/ADRD (e.g., resistant) and vulnerable regions. Knowing that age is the prime risk factor for the most prominent neurodegenerative disorders in humans, the effect of age-related changes and the senescence status of different brain cell types need to be understood in more detail.
The Cellular Senescence Network (SenNet) is a collaborative research effort launched by the NIH Common Fund to comprehensively identify and characterize the heterogeneity of senescent cells in multiple human (and other model organism) tissues, in various states of health, and across the lifespan. Ultimately, the network aims to develop public atlases of these cells and create tools and technologies (e.g., biomarker panels, computer and experimental model systems, imaging tools) that build from other Common Fund programs to advance the development of therapeutics (NIH, 2024; SenNet Consortium, 2022).
Epigenetic Dysregulation
DNA methylation in the brain trends toward global decreases with aging, which has been associated with enhanced gene expression (De Jager et al., 2014; Pellegrini et al., 2021). The pattern and frequency of epigenetic changes—in particular, DNA methylation at cytosine-guanine pair sites—have been proposed as a mechanism for comparing chronological and biological age (i.e., an epigenetic clock) (Gonzales et al., 2022). Differential methylation patterns have been observed in genes associated with AD, such as APP and MAPT, among others, when comparing individuals with AD to control subjects (De Jager et al., 2014; Iwata et al., 2014; Yu et al., 2015). As age advances, increases in chromatin remodeling and chromatin heterogeneity are also observed. In addition to DNA methylation, histone acetylation typically decreases with aging, resulting in a chromatin structure that is more condensed and consequent changes in transcription (Gonzales et al., 2022; Peleg et al., 2016). By analyzing postmortem human brain tissue, a recent study uncovered an increase in the expression of two histone acetyltransferases, which were associated with both amyloid beta pathology and neurodegeneration (Nativio et al., 2018, 2020).
Alterations in levels of multiple mitochondrial RNAs have been found in AD cases, which may play a role in multiple aspects of pathology, such as modulating amyloid beta and tau production and function, synaptic
plasticity, neuronal growth, apoptosis, and inflammatory response. Mitochondrial RNAs may therefore be useful as potential biomarkers and therapeutic targets (Zhang and Bian, 2021).
Understanding the relationships between the exposome, age, epigenetic and posttranslational changes, and such age-related diseases as AD/ADRD holds promise for disease prediction (Grodstein et al., 2020; Shireby et al., 2020) and intervention efforts. As potentially reversible contributors to AD/ADRD, epigenetic and posttranslational alterations are appealing targets for therapeutic interventions (Hara et al., 2019; Migliore and Coppedè, 2022). Realizing such potential will require the systematic analysis and comparison of epigenetic and posttranslational changes in aged and diseased (AD/ADRD) brain tissue and identification of areas of overlap.
Toward an Integrative Biological Framework for AD/ADRD
As illustrated in Figure 3-3 above and described throughout this section, the process of neurodegeneration is influenced by a number of cell types and pathogenic processes. Importantly, the factors and mechanisms affecting the onset and progression of disease act along the life course, meaning they occur to different extents as the brain ages, and may also interact with each other. These processes (e.g., inflammation, vascular dysfunction, altered proteostasis and autophagy, mitochondrial and metabolic changes) and cellular players (e.g., neurons, endothelial and glial cells) comprise a complicated biological framework for neurodegeneration. It is increasingly acknowledged that age-associated neurodegenerative diseases, such as AD/ADRD, are the result of complex and multifactorial events. However, all the pathways described are disease relevant and need to be taken into account when creating a more integrative understanding of events taking part in age-related neurodegeneration (Behl, 2023; Lehmann et al., 2023).
By enabling better discernment of the initial triggers, cascading pathophysiologic sequences, and biological systems working to counteract those perturbations to restore homeostasis, such a model could potentially guide combination approaches to AD/ADRD interventions that sequentially target key steps in the etiologic cascade and that integrate age-dependent changes. For example, nonpharmacological interventions implemented early in life for individuals at risk for dementia (e.g., based on genetic and exposome risks) could be followed later in life with an anti-inflammatory therapy, and potentially, a pathology-specific (e.g., amyloid) immunotherapy to avoid microglia activation. Such a combination approach may be necessary to achieve effect sizes greater than those observed to date for strategies targeting a single pathology and pathway.
Some progress has been made in the development of integrated models for molecular and cellular pathways contributing to neurodegeneration
(Balusu et al., 2023; De Strooper and Karran, 2016; Gan et al., 2018). In such models, a biochemical phase features perturbations in normal cellular processes that contribute to the protein aggregation commonly observed in neurodegenerative diseases; protein aggregation, on the other hand, is not part of the pathological process in dementia types based on vascular changes (vascular dementia). The biochemical phase is followed by a cellular phase during which cells in the brain react to the protein aggregates. A key element of the cascade proposed here is the generation of an innate inflammatory reaction mediated by microglia, which triggers reactions from other cells and ultimately leads to neuronal cell death. The resulting damage to the brain, some potentially irreversible, leads to the clinical phase (cognitive impairment and clinical dementia) (De Strooper and Karran, 2016; NASEM, 2024).
While described linearly for simplicity, within this cascade there are several feedback mechanisms, adding to the model complexity, as depicted in Figure 3-3 earlier in this chapter. On the other hand, this proposed sequence leaves behind the neuron-only modeling of neurodegeneration and considers also the effect of other nonneuronal cell types. As neuroinflammatory endophenotypes and biomarkers for different biochemical pathways are developed, it may be possible to further unravel the temporal relationship between inflammatory responses and other pathophysiological aspects of AD/ADRD (Arafah et al., 2023; Hampel et al., 2020).
While the aforementioned cascade provides a useful starting place for understanding the interdependent molecular and cellular processes contributing to AD/ADRD, a comprehensive model needs to account not only for pathologic factors occurring within the brain, but also the role of the exposome, brain–peripheral body interactions, and mechanisms of resilience.
The Role of the Exposome and Brain–Peripheral Body Interactions
Upstream of the intracellular biochemical events leading to the different pathogenic processes are the contributions of the exposome to pathogenesis, which need to be incorporated into an integrative model. While it is clear that genetic variants associated with AD/ADRD can increase susceptibility to and/or accelerate many of the hallmarks of aging, the contributions of exposome factors, mediated either through gene–environment interactions or through direct biochemical and/or cellular effects, have not been well characterized and are important for understanding differences in AD/ADRD across diverse populations. As discussed earlier in this chapter, efforts are needed to identify exposure-related pathways that modulate or converge with other AD/ADRD risk factors or pathways to mediate pathogenesis. For example, air pollution may alter the blood–brain barrier (Oppenheim et al., 2013) and has also been shown to reduce levels of
esterified lipid mediators, which, as noted earlier, are involved in resolving inflammation and have been found to be deficient in AD (Shen et al., 2022; Taha, 2024), indicating the potential for synergistic deleterious effects.
A growing body of evidence suggests that infections, both CNS specific and systemic, can increase the risk of dementia and that these effects may be mediated in part by inflammation, although pathogen-specific pathways may also be involved (Itzhaki, 2021; Sipilä et al., 2021; Vestin et al., 2024), particularly for pathogens that can infect the brain, such as herpes simplex virus (Itzhaki, 2021) and SARS-CoV-2 (Liu et al., 2023b). Furthermore, behavioral factors such as diet and physical activity contribute to cardiovascular risk factors, such as hypertension, diabetes, and obesity (Gonzalez et al., 2024; Marquez et al., 2024) and are increasingly understood to contribute to neurodegeneration through vascular pathways (NASEM, 2018), but behavioral factors may also affect AD/ADRD risk through epigenetic effects (Maloney and Lahiri, 2016; Migliore and Coppedè, 2022).
The potential for extra-CNS infections to increase dementia risk and the well-established cardiovascular risk factors for dementia underscore the need for brain–peripheral body interactions to be represented in a more holistic integrated model (Hampel et al., 2019). AD/ADRD are not diseases exclusive to neurons but involve the whole body since the brain and the (rest of the) body are tightly connected through metabolic, immune, and vascular systems (Hampel et al., 2019). As discussed earlier in this chapter, vascular dysfunction may be linked to such neurodegenerative mechanisms as neuroinflammation and impeded clearance of protein aggregates. Thus, targeting cardiovascular risk factors in the periphery (e.g., atherosclerosis, hypertension) has the potential to affect multiple potential AD/ADRD pathways in the brain in a disease-agnostic manner, in addition to effects on cardiovascular diseases such as heart disease and stroke (preventive interventions targeting cardiovascular risk factors are discussed in Chapter 4).
Another notable example of such brain–peripheral body interactions is the gut microbiome and its potential contribution to AD/ADRD through the gut–brain axis. Microbial metabolites may exert effects on the brain through multiple pathways (e.g., immune, endocrine, enteric nervous system), and are thought to have a wide range of effects including altering neuronal transcription, compromising the blood–brain barrier integrity, and triggering neuroinflammation (Wang et al., 2021). Ongoing research is seeking to elucidate how gut microbiota interact with certain risk gene settings and exposome factors, such as air pollution (Dutta et al., 2022), to influence the onset and/or progression of AD/ADRD. In addition to the gut–brain axis, connections via the lung–brain and renal–CVD–brain axes have also been studied (Finch and Kulminski, 2019; Owaki et al., 2024).
The Role of Cognitive Resilience
Also critical for inclusion in an integrated model are pathways that provide an explanation for why many individuals with evidence of accumulated brain pathologies and neurodegenerative changes fail to develop cognitive impairment or clinical dementia (Aizenstein et al., 2008) and why others do not develop neuropathology, as can be observed in a cohort of centenarians (Zhang et al., 2023e). As an example, one-third of individuals in the Religious Orders Study and Rush Memory and Aging Project (ROSMAP) cohorts who met the diagnostic criteria for pathologic AD at the time of autopsy had no clinical signs of cognitive impairment at the time of death, suggesting the existence of resilience factors—endogenous (e.g., personality, life outlook, genetic factors) and/or exogenous (e.g., access to resources)—capable of offsetting the detrimental effects of brain pathologies (i.e., a cognitive reserve hypothesis) (Katzman et al., 1988; Zammit et al., 2023).
Genomics and other omics approaches are helping to identify resilience factors, elucidate their underlying biological mechanisms, and to relate them to neuropathology and changes in brain function (Box 3-3 describes the Resilience-AD program that is supporting some of this work) (de Vries et al., 2024). Proteomics studies using samples from ROSMAP cohorts have shown that synaptic function and mitochondrial activity are enriched by proteins associated with higher cognitive stability, while proteins associated with more rapid cognitive decline were enriched for inflammatory response, apoptosis, and endothelial function in glial cells (Zammit et al., 2023). Of interest, some proteins associated with cognitive resilience are pleiotropic, also conferring resilience to motor phenotypes of neurodegenerative diseases, underscoring the breadth of health benefits afforded by resilience factors. While specific mechanisms of action are still being uncovered, resilience factors may fall into three different mechanistic categories: (1) those affecting clinical phenotypes independent of brain pathologies, (2) those that interact with brain pathologies to attenuate or modify their effects, and (3) those directly affecting the magnitude of brain pathologies (i.e., conferring resistance to pathology) (Zammit et al., 2023).
The identification of modifiable resilience factors can guide the development of preventive and therapeutic approaches for AD/ADRD (Neuner et al., 2022). For instance, evidence regarding the contributions of physical activity and education to resilience provide support for individual-level and public health nonpharmacologic interventions focused on these factors (discussed further in Chapter 4). Moreover, advancing understanding of the biological mechanisms underlying cognitive resilience can inform drug discovery. For example, increased levels of Neuritin 1, a protein with an important role in plasticity and synaptic function, is associated with cognitive resilience
BOX 3-3
Resilience-AD
The Resilience-AD program was established with the goal of improving understanding of how gene–environment interactions contribute to cognitively resilient phenotypes in people at high risk for Alzheimer’s disease (AD), with the ultimate objective of identifying novel therapeutic targets for pharmacological and nonpharmacological interventions aimed at prevention (AD Knowledge Portal, 2024b). Resilience-AD is intended to be complementary to and inform other efforts focused on the discovery of novel targets and biomarkers, including the Accelerating Medicines Partnership® Program for Alzheimer’s Disease and M2OVE-AD Programs. The program will generate multiomic (e.g., genomic, proteomic, metabolomic) profiling data from samples collected from individuals who (1) are resistant to the development of AD/ADRD neuropathology despite high genetic risk (e.g., APOE4 homozygotes, people with Down Syndrome, autosomal dominant mutation carriers), (2) reach very old age (90 years of age or older) without developing dementia, or (3) remain cognitively normal despite the presence of neuropathologic disease biomarkers (AD Knowledge Portal, 2024b). Using network biology approaches, molecular, clinical, pathologic, and exposome (e.g., environmental, lifestyle, experiential) data collected from individuals exhibiting resilience to AD will be integrated and used to identify molecular networks that can predict cognitive resilience.
and has been identified as a therapeutic target for AD (Hurst et al., 2023; Zammit et al., 2023). Cellular studies aiming to define resistance and adaptive factors that provide protection against neurodegeneration-associated challenges and stress (e.g., oxidative and endoplasmic reticulum stress) may complement in vivo investigations (Chakraborty et al., 2019; Pham et al., 2023). To advance precision approaches to AD/ADRD interventions, resilience-focused research and an integrative biological framework for AD/ADRD need to consider the interplay of biological and sociocultural factors and the consequent potential for heterogeneity in resilience determinants and trajectories across different population groups. An important example is the observation of sex and gender differences in resilience, which may stem from myriad interrelated factors requiring further study, including neuroanatomy, genetic architecture, and influences of gender roles on modifiable risk factors (Arenaza-Urquijo et al., 2024).
Targeting resilience factors represents an appealing alternative to targeting individual AD/ADRD pathologies, especially given the prevalence of mixed pathologies. In the future, a cognitive resilience index
based on the ability to measure known resilience factors could facilitate a precision medicine approach to interventions, which may include combinations of resilience behaviors, such as physical activity, and novel resilience therapies aimed at maintaining brain health and late-life cognitive function.
Conclusion 3-2: AD/ADRD has complex multifactorial origins. Fundamental gaps in the current understanding of the underlying biology of dementia and the siloed study of neurodegenerative diseases and mechanistic pathways impede efforts to develop effective prevention and treatment strategies. The investigation of pathways and mechanisms that are shared across neurodegenerative diseases will provide opportunities to identify targets for prevention and treatment that are not specific to a singular pathology.
Conclusion 3-3: The creation of a comprehensive biological framework that integrates knowledge of the shared molecular and cellular mechanistic pathways underlying neurodegenerative disease and their interplay with exposome, genetic, and resilience factors, among others, would provide a holistic model that could facilitate the translation of basic research into effective preventive and therapeutic interventions across multiple pathologies. Resilience and resistance factors that provide normal cognitive functions despite disease-like pathologies need to be uncovered.
Model Systems and Other Tools for Elucidating Molecular and Cellular Pathways Leading to AD/ADRD
Considering the various and interrelated molecular and cellular pathways discussed in the sections above, one central question remaining is how to model complex brain diseases that develop over decades and are affected by various sets of risk genes, as well as their interplay with peripheral organs (i.e., system health) and the environment (i.e., exposome). NIH has made substantial investments to support the creation of model systems to serve as an experimental basis for studying disease pathogenesis and facilitating drug discovery. As no single model is able to fully recapitulate the human condition, and each has its strengths and weaknesses, a variety of in vivo and in vitro models have been developed to answer different kinds of scientific questions. Thus, it is important to adequately define the key questions at the start of a scientific endeavor so they may drive the development and selection of an appropriate model. In the understandable absence of an ideal model, the use of multiple models in combination can enhance the robustness of the experimental findings.
As with other tools, all models need to be adequately validated to ensure reliability and reproducibility for a given context of use (i.e., how and when the model will be used). The stringency of the validation process depends on how the data will be used (NASEM, 2023). For example, different levels of stringency would be needed for validation of models used to understand mechanistic pathways through basic research versus those used to inform decisions regarding the safety of a novel therapeutic.
In Vivo Models
While AD/ADRD are explicitly human disorders and do not appear to occur naturally in conventional laboratory rodents, genetic engineering has enabled the development of mouse and rat models for use in elucidating pathogenic processes that lead to different types of neuronal dysfunction and evaluating potential therapeutic approaches (Sinclair et al., 2024). For AD alone, there are hundreds of mouse models available and each may have different trajectories and timelines related to the formation of plaques, tangles, neuronal dysfunction, gliosis, synaptic loss, and memory impairment (AlzForum, 2024). These mouse models are mostly based on amyloid and tau biology. Despite limitations stemming from differences in human and murine anatomy and physiology and potential artifacts from the genetic engineering process, mouse models have also played an important role in enhancing understanding of the role of various genes, such as APOE4 (Padmanabhan and Gotz, 2023) or those encoding elements of the endosomal–lysosomal system (Lee et al., 2022; Terron et al., 2024) in AD/ADRD, as well as pathogenic processes such as the prion-like neuron-to-neuron spreading of amyloid and tau pathology (Padmanabhan and Gotz, 2023).
While many transgenic mouse models were generated using genetic alterations associated with dominantly inherited disease forms (e.g., familial AD and FTD), concerns regarding the applicability of such models to sporadic, late-onset disease forms and their failure to predict the clinical efficacy of therapies in clinical trials spurred efforts to create novel animal models for late-onset disease using genetic engineering methods such as CRISPR and other innovative approaches (Collins and Greenfield, 2024; Oblak et al., 2020; Padmanabhan and Gotz, 2023). One prominent NIA-funded effort is the Model Organism Development and Evaluation for Late-onset Alzheimer’s Disease (MODEL-AD),8 which is using computational analyses of human datasets made available from other NIH initiatives such as the Accelerating Medicines Partnership® Program for Alzheimer’s Disease (AMP-AD), M2OVE-AD, ADSP, ADNI, and other sources to guide the development of novel models that recapitulate key hallmarks of late-onset
___________________
8 Further information is available at https://www.model-ad.org/ (accessed June 5, 2024).
AD. Ultimately, the aim is to integrate these models into preclinical testing pipelines to identify and evaluate novel therapies (MODEL-AD, 2024; NIA, 2023b). MODEL-AD, like most animal model development to date, has primarily focused on AD, with a heavy bias toward amyloid pathways, although some models for LBD (Lim et al., 2011; Magen and Chesselet, 2011; Sander et al., 2023; Stylianou et al., 2020) and FTD (Ahmed et al., 2017; Kashyap et al., 2023; Roberson, 2012) have been created. To continue to expand the therapeutic pipeline to other nonamyloid targets and related dementias, there is a need for further investment in the creation and characterization of models that can inform those efforts, including mixed-pathology models. Animal models are also critical to understanding synaptic and cellular contributors to resilience (de Vries et al., 2024; Neuner et al., 2022).
In addition to mice, other animals with the potential to model AD/ADRD include fruit flies (Trotter et al., 2017); rats; dogs, which naturally develop amyloid plaques with age (Noche et al., 2024); and nonhuman primates (NHPs) (Padmanabhan and Gotz, 2023). Rats are commonly used in pharmacokinetic studies but also may be superior to mice in modeling inflammation (Du et al., 2017; Seok et al., 2013) and complex cognitive behaviors. NIA’s intramural research program is conducting a longitudinal study of neurocognitive aging in rats—Successful Trajectories of Aging: Reserve and Resilience in RatS (STARRRS)—which will generate shareable resources of phenotypic and neuroimaging data, as well as biological samples (NIA, 2024c)
As compared to rodents, NHPs are more similar to humans with regards to neuroanatomy and neurophysiology (NASEM, 2023), which may make them useful models for neurodegenerative diseases such as AD/ADRD. Some NHPs (great apes, macaques) develop amyloid plaques with age but they do not appear to regularly spontaneously develop other features of human AD pathology that lead to dementia (e.g., neurofibrillary tangles) (Beckman et al., 2021). Consequently, there is interest in genetically engineering more human-relevant NHP models for AD/ADRD. NIH recently funded the Marmosets As Research Models of AD (MARMO-AD) program, which is exploring transgenic marmosets as models to study primate‐specific cellular and molecular mechanisms that contribute to the pathogenesis and progression of AD (Sukoff Rizzo et al., 2023). Genetic engineering of new NHP models is still in its infancy, however, and may be slowed by the high cost and logistic hurdles of working with NHPs, as well as ethical considerations (Padmanabhan and Gotz, 2023).
Consistent with NIH policy requirements (NIH, 2015), NIH-funded AD/ADRD studies using animal models need to consider sex as a biological variable, beginning with the development of research questions and extending to analysis of results and reporting of findings (e.g., sex disaggregated).
In doing so, preclinical animal studies can help to improve understanding of sex-specific differences in AD/ADRD (Arenaza-Urquijo et al., 2024).
In Vitro Models
To complement AD/ADRD research using animal models, there has been increasing focus on the development of in vitro models derived from human cells in the hopes of generating more human-relevant findings. Cell-based systems for studying human diseases were revolutionized in the early 2000s by the discovery of human induced pluripotent stem cells (iPSCs) (Yamanaka, 2012), which can be used to generate most human tissue cell types. Patient-derived iPSCs have been used to develop human brain organoids and brain microphysiological systems (i.e., tissue chips), which are being explored as potential model systems to study AD/ADRD. The generation of iPSCs from participants involved in cohort studies provides opportunities to link data from the cell-based models to longitudinal clinical data (NIA, 2022a).
Additional and further advanced in vitro systems that can recapitulate patient-like pathophysiology are emerging and represent potential alternatives to conventional animal- and cell-based models (Amartumur et al., 2024). For example, brain or cerebral organoids are three-dimensional (3D) cellular aggregates that when generated from human iPSCs exhibit some similarities to the structural and functional features of specific regions of a human brain, including neural circuit formation and neuroimmune and neurovascular interactions. Such models have been used to compare amyloid clearance by microglia carrying the APOE4 or the APOE3 allele (Paşca, 2019) and to elucidate early pathogenic events resulting from mutations in the MAPT gene that gives rise to an inherited form (autosomal dominant) of FTD (Bowles et al., 2021). As their architecture and functionality more closely resemble an embryonic brain, brain organoids may generally be better suited to the study of neurodevelopment than age-related neurodegeneration (Paşca, 2019).
Microphysiological systems are similarly 3D cellular systems intended to recapitulate specific aspects of human physiology but differ from organoids in that they are formed on microfluidic chips, giving researchers more control over the development of the composition and architecture of the tissue and the ability to model such features as vascular perfusion (Hargrove-Grimes et al., 2021). Coculture systems that can enable the investigation of complex cellular interactions (e.g., among neurons, astrocytes, microglia, endothelial cells) (Cetin et al., 2022) have the potential to help answer pressing scientific questions regarding the perturbations in cellular activity that lead to the loss of homeostasis in the brain (De Strooper and Karran, 2016). For some scientific questions regarding AD/ADRD biology, such coculture systems
may better model in vivo conditions and have greater translational relevance than cellular models involving only a single cell type (Cetin et al., 2022). Furthermore, the ability to connect chips with different tissues in a modular fashion can enable the study of interactions of peripheral body organs with the brain. A workshop hosted by NIH in 2022 focused on opportunities for microphysiologic systems to serve as alternatives to animal models for AD/ADRD and their potential role in drug discovery and development (NIA, 2022a). This workshop highlighted examples of efforts underway to develop 3D cell-based models for AD/ADRD including an organoid model for AD and tissue chip models for FTD and the blood–brain barrier.
An important limitation of iPSC-derived organoid and microphysiologic models is that the process of reprogramming patient-derived cells into iPSCs results in the loss of their somatic epigenetic patterns, including aging, exposure, and disease-related markers (Balusu et al., 2023). A promising complementary approach that may circumvent this limitation is induced neuron (iN) models, which are generated by converting already differentiated cells, commonly fibroblasts, into neurons (Balusu et al., 2023; Traxler et al., 2022). In contrast to iPSC models, iN models retain to some degree (albeit not well characterized) the epigenetic profile and metabolic state of the patients they are derived from and demonstrate enhanced expression of mature neuronal markers and improved functionality (Balusu et al., 2023). Employing iN models may lead to a better understanding of neuronal state and fate changes, which in turn could guide intervention approaches to manipulation of cell fate with the goal of promoting neuronal resilience, repair, and, potentially, rejuvenation (Traxler et al., 2023). Further comparative evaluations of iPSC and iN models are needed to better characterize the advantages and limitations of each and their use as complementary systems.
Of course, in vitro models are not suited to some aspects of AD/ADRD research, such as elucidating the underlying effects of such factors as exercise, hypertension, psychological stress, sociobehavioral factors, and sleep, which generally require the study model to be an integrated organism. Other considerations regarding findings from human-derived cell models include potential variability resulting from such factors as differences in cell sources, genetic backgrounds, and culture methods (including their ability to mimic physiologic conditions) (Slanzi et al., 2020), as well as how representative existing models are of the population and different dementia phenotypes.
Single-cell sequencing of samples from brains collected postmortem from individuals with AD/ADRD have the potential to generate data that are complementary to cell-based models, such as uncovering affected neuronal and nonneuronal cell types, vulnerable brain regions, and changes (e.g., somatic DNA mutations, altered gene expression) that precede neuronal dysfunction and death (Balusu et al., 2023; Miller et al., 2022). Advances in
multiomics approaches are enabling the analysis of genomic, transcriptomic, metabolomic, lipidomic, and epigenomic information from single cells (Vandereyken et al., 2023), providing new opportunities to elucidate molecular pathways leading to dementia, especially when coupled with emerging spatial multiomics methods. In addition, multiomics approaches in combination with GWAS and genetic single-nucleotide polymorphism studies have the potential to define “a cluster of core alterations in AD” and can advance a more holistic approach in AD/ADRD research (Argyriou et al., 2024). As discussed later in this chapter, findings from studies using unbiased GWAS and multiomics approaches require significant investment in follow-on basic, mechanistic research (including studies using in vivo models) to understand their biology and how they might be targeted therapeutically.
One challenge with the high-dimensional analyses of single cells from postmortem brain samples is that the disease has generally already progressed to late or end stage and data can only be captured from the more resistant, surviving neurons. To address these limitations, data from single-cell analyses of human brain samples could be supplemented with single-cell multiomics data from animal models, which may provide insights into earlier-stage dysfunction (Balusu et al., 2023). Of note, the Brain Research Through Advancing Innovative Neurotechnologies® (BRAIN) initiative recently released a comprehensive single-cell spatial map of the mouse brain and identified thousands of cell types, underscoring the importance of leveraging large-scale resources developed by other institutions.
Conclusion 3-4: Animal and in vitro models have important roles to play in enhancing understanding of select molecular, biochemical, and physiological processes of AD/ADRD. While no model can recapitulate the full complexity of disease pathogenesis across the human life course, preclinical research can elucidate elements of this complexity and links between pathogenic factors and risk genes.
ACCELERATING THE TRANSLATION OF BASIC, EPIDEMIOLOGICAL, AND SOCIAL SCIENCES RESEARCH FOR THE DEVELOPMENT OF INTERVENTION STRATEGIES
As described in this chapter, diverse streams of epidemiological, social science, and mechanistic research have been pursued, funded by NIH and others (Stephan et al., 2024). An expansion beyond this breadth of inquiry is appropriate given the breadth of the exposome and the longitudinal and molecular complexity of pathways that lead to AD/ADRD. All of these factors contribute to the individuality of this group of neurodegenerative diseases, as increasingly supported by recent advances in biomarkers and subtyping studies (Tijms et al., 2024), as discussed further in Chapter 4.
Advances in the understanding of the risks associated with different types of dementia and the molecular and cellular pathways that mediate those risks need to be accompanied by efforts to translate this new knowledge into intervention strategies. These efforts can be guided by advancements in blood-based or other biomarkers to stratify risks into all-cause dementia, AD, or related dementia risks.
Translational Epidemiology Approaches: Moving from Risk Factors to Public Health Interventions and Back to Stratified Epidemiology
Research to guide interventions must evaluate causation, and yet for decades the statistical training typically offered to clinical researchers has emphasized “risk factor” epidemiology (i.e., the identification of correlates of disease without rigorous evaluation of causality). Revolutionary progress in conceptualization and methodology for causal inference started in the 1980s and created substantial debates in clinical research (Pearl, 2009). Technical tools to support causal inference in the absence of formal randomization continue to improve, alongside rigorous frameworks for recognizing the limitations of evidence from randomized and nonrandomized studies (Hernán, 2018). This progress is highly relevant to AD/ADRD research and yet very few researchers have adopted these tools. The slow adoption is attributable to a combination of statistical difficulty, the limited formal training available to many researchers (the field is deeply interdisciplinary), or training that occurred prior to the causality revolution and thus did not incorporate the tools. As a result, the quality of evidence available even for routine questions that have been tackled in countless studies, such as the effects of alcohol use on AD/ADRD, or common medications, remains thin.
Many observational studies are vulnerable to identical biases, so they offer little new evidence to complement one another. More formal frameworks to integrate evidence across study designs, populations, and data types are needed. This includes work on measurement harmonization and cross-walking, meta-analyses and meta-regression, and evidence triangulation (Lawlor et al., 2016; Thurmond, 2001). For example, randomized controlled trials are not always or even often feasible, especially for preventive interventions for a disease that develops over decades. As a result, conclusions about preventive strategies rest heavily on studies that were not intentionally designed randomized controlled trials.
There are two general strategies for evaluating causality in these settings: (1) control for confounders that might influence the exposure and also AD/ADRD or (2) identify a quasi-random source of variation in exposure and evaluate how that influences AD/ADRD risk (Matthay et al., 2020). Combining evidence from both approaches offers a far stronger basis for causal conclusions. Additional opportunities for evidence triangulation include
identifying settings where the confounders are likely to be entirely different or finding negative control outcomes. These approaches align well with the policy emphasis in Chapter 4, highlighting the strategies of public and institutional policies to translate epidemiological findings into public health impact for prevention of AD/ADRD. Policies are often directly amenable to evaluation via quasi-experimental approaches, for example, leveraging approximately random variation in the timing of policy adoption and implementation. Major progress already achieved in declining age-specific incidence rates in many settings suggest rigorous evaluation of the changes that account for those declines may point the way to future breakthroughs.
In addition, as blood tests for AD/ADRD are implemented in real-world settings, the availability of biomarker data will enable epidemiological research to stratify the population and differentiate risk factors for AD from those for related dementias. This will enable the development of tailored interventions in both public health and therapeutics.
Conclusion 3-5: Despite substantial evidence, the translation of epidemiological research to public health interventions has been slow and has been hindered by insufficient evaluation of causation that can be addressed, in part, through intensive statistical work to triangulate evidence from different study designs, types of data, and populations.
Opportunities to Accelerate Discoveries into Interventions for AD/ADRD
At present there are more than 100 drugs being evaluated in clinical trials for effectiveness against AD (Cummings et al., 2023b), many targeting pathways not specific to any one AD/ADRD pathology, such as those described earlier in this chapter. More than 150 trials are evaluating nonpharmacological interventions (Kelley, 2023). Despite hope for each approach, the reality is that most phase 2 trials will fail. Thus, improving our ability to learn from failures, as well as improving the probability of success for the pharmacological interventions entering the clinic, needs to be a research priority as it has the greatest potential to improve the delivery of treatments for AD/ADRD. Negative clinical trials need to be analyzed to identify points of failure in translation and the potential causes, and this information needs to be fed back into earlier phases of the research and development pipeline, as discussed further in Chapter 4. However, this can only occur if trials are well designed and have specific and validated target engagement or pharmacodynamic biomarkers informing upon the hypothesized mechanism of action. Currently only 44 percent of clinical trials have fluid biomarkers fulfilling this role and the numbers drop in industry-sponsored trials (Marlies Oosthoek, 2024), and even fewer have imaging measures because of the associated costs.
This is a significant barrier to the translation of clinical research back to basic mechanisms.
Success of clinical drug discovery research can be seen two ways. First, success is achieving a positive primary outcome providing evidence of efficacy on an accepted endpoint. This type of success leads to phase 3 studies to demonstrate application across a larger and more diverse patient population. However, this does not occur often; instead, many drug discovery trials fail to meet their primary outcome. Outside of safety, the most important secondary outcome is that the intervention engages the mechanism and provides sufficient activity such that an efficacy response would have been observed if that mechanism was related to the disease. The majority of drug discovery trials do not invest sufficiently in this area and thus, despite the investment in clinical research, the field advances slowly and leaves many questions pertaining to why efficacy was not achieved.
Conclusion 3-6: Almost all early-phase clinical research (phase 1b and 2 trials) fails to achieve its primary efficacy outcomes. However, the return on the investment in clinical research can be dramatically improved through the focus on testing specific mechanistic hypotheses and employing specific and analytically validated measures in these early-phase trials. Where this is not yet possible, biobanking can facilitate future understanding if samples are easily accessible by academic researchers. Such an approach will improve the quality and value of the results obtained and ensure that the participants’ involvement in drug trials achieves maximum benefit.
The probability of success for pharmacological interventions entering the clinic is low and requires improvements in target diversity, validation, and enablement. NIH has intensified infrastructure investments in these areas and has developed several interconnected translational research programs that support the identification, validation, and enablement of targets as part of a precision medicine approach to drug development for AD (see Figure 3-6) (NIA, 2023b). These strategic investments in the earliest phases of drug discovery and preclinical research have resulted in the generation of large and diverse (e.g., genetic, exposome, functional multiomics) datasets that can be used for the identification of novel targets.
The Accelerating Medicines Partnership® (see Box 3-4), a cross-sector partnership launched in 2014 and managed by the Foundation for the NIH has used several avenues to identify pathways and nodes specific to certain cell types that are affected in AD (NIA, 2024d). The Target Enablement to Accelerate Therapy Development for Alzheimer’s Disease (TREAT-AD) centers function to bridge the efforts of AMP-AD and those to enable novel targets through early drug discovery using animal models developed
NOTE: ABC-DS = Alzheimer Biomarkers Consortium–Down Syndrome; ACTC = Alzheimer’s Clinical Trails Consortium; ADNI = Alzheimer’s Disease Neuroimaging Imitative; ADSP = Alzheimer’s Disease Sequencing Project; AMP-AD = Accelerating Medicines Partnership—Alzheimer’s Disease Target Discovery and Preclinical Validation; CLEAR-AD = Centrally-linked Longitudinal pEripheral biomARkers of Alzheimer’s Disease; IND = investigational new drug; TREAT-AD = Target Enablement to Accelerate Therapy Development for Alzheimer’s Disease; HABS-HD = Health and Aging Brain Study—Health Disparities; MARMO = Marmosets as Research Models for Alzheimer’s Disease; MODEL—AD = Model Organism Development and Evaluation for Late-onset Alzheimer’s Disease.
SOURCE: NIA, 2024e.
BOX 3-4
The Accelerating Medicines Partnership®
The Accelerating Medicines Partnership® (AMP) program is a public–private partnership that brings together federal agencies (NIH, FDA), industry (e.g., biopharmaceutical and life sciences companies), and nonprofit organizations to reduce the time and costs associated with making new diagnostics and treatments available to patients by fostering joint efforts to identify and validate promising biological targets for therapeutics (NIA, 2024d). The cross-sector partnership was launched in 2014 and is managed through the Foundation for the NIH (FNIH). Several disease-specific AMP programs have been established, including but not limited to programs for AD (NIA, 2024d), PD (AMP-PD, n.d.), and ALS (NINDS, 2024b). Given the potential for commonalities and overlapping mechanisms among these neurodegenerative diseases, coordination will be important going forward to ensure siloing of efforts does not result in missed opportunities to leverage advances in knowledge, innovation, and infrastructure and to foster a more integrative approach.
Accelerating Medicines Partnership® Program for Alzheimer’s Disease (AMP-AD)
Initiated in 2014, AMP-AD is part of a larger NIH effort to identify and validate novel biomarkers and therapeutic targets for AD (e.g., in conjunction with CLEAR-AD and TREAT-AD) (Kelley, 2023). All AMP-AD partners have agreed to making the data and analyses from this initiative publicly available for broad scientific use; these data can be accessed via a centralized data infrastructure and data-sharing platform, the AD Knowledge Portal, and through the open-source platform, Agora (NIA, 2024d). In its first iteration, AMP-AD was focused on identifying novel therapeutic targets and evaluating tau imaging as a biomarker for measuring disease progression and response to treatment. The second iteration, AMP-AD 2.0, was launched in 2021 to expand on the prior efforts to discover and validate new targets and biomarkers using a precision medicine approach (NIA, 2022b). Supported by a series of NIH foundational grants, specific strategic directions for AMP-AD 2.0 include the following:
- “Expand the molecular profiling in brain, CSF, and blood samples from diverse cohorts
- Generate longitudinal immunologic profiling data to enable dynamic modeling of the disease trajectory
- Expand the existing single-cell/single-nucleus molecular profiling efforts to build predictive models of the disease at single-cell resolution” (NIA, 2022b).
Accelerating Medicines Partnership® Program for Parkinson’s Disease and Related Disorders (AMP-PDRD)
The Accelerating Medicines Partnership® Program for Parkinson’s Disease (AMP-PD) was established in 2018 as a partnership between NINDS, NIA, FDA, several industry partners, and the Michael J. Fox Foundation for Parkinson’s Research (AMP-PD, n.d.). AMP-PD aims to identify and validate biomarkers (diagnostic, prognostic, and progression) biomarkers with the longer-term objective of improving clinical trial design and advancing the identification of new pathways for development of therapeutics. In pursuit of this goal, AMP-PD leverages samples and data from existing well-characterized cohorts including the NINDS Parkinson’s Disease Biomarkers Program, the BioFIND study, and the International Lewy Body Dementia Genetics Consortium Genome Sequencing in Lewy body dementia case-control cohort, among others (AMP-PD, n.d.). Building on the work of AMP-PD, AMP-PDRD was initiated by FNIH in 2024 with the goal identifying and validating biomarkers that can be used to distinguish PD from related disorders with similar biological characteristics and symptoms (e.g., LBD, multiple system atrophy, progressive supranuclear palsy). Ultimately these efforts are intended to improve the success of clinical trials, expand therapies for these related conditions, and enable precision medicine approaches to treatment (FNIH, 2024).
through MODEL-AD (see Box 3-5). The Centrally Linked Longitudinal Peripheral Biomarkers of Alzheimer’s Disease in Multiethnic Populations (CLEAR-AD) program is exploring through a diverse set of samples centrally linked and longitudinal peripheral biomarkers to enable personalized medicine approaches (see Box 2-6). Coupled with the investments in the Alzheimer’s Disease Research Centers, these programs function to derisk novel targets and facilitate their translation toward clinical development (NIH, 2019) or the Small Business Innovative Research program. Considering the comparatively small number of clinical trials for dementia relative to other high-burden diseases such as cardiovascular disease and cancer (see Chapter 1), these programs are critical to enabling the expansion of early-phase trials of preventive and therapeutic interventions for AD/ADRD, as discussed further in Chapter 5.
However, the importance of considering both AD and related dementias cannot be overstated. As the field has expanded beyond amyloid-targeting mechanisms, investigators have found that the mechanistic hypotheses for
BOX 3-5
Target Enablement to Accelerate Therapy Development for Alzheimer’s Disease (TREAT-AD)
Two complementary TREAT-AD drug discovery centers—one led by researchers at the Indiana University School of Medicine and Purdue University, and the second by researchers at Emory University, Sage Bionetworks, and the Structural Genomics Consortium (NIA, 2023c)—were established with a mission to “improve, diversify and reinvigorate the Alzheimer’s disease (AD) drug development pipeline by accelerating the characterization and experimental validation of next generation therapeutic targets and integrating the targets into drug discovery campaigns” (TREAT-AD, 2024). NIH support for the two centers is estimated at over $73 million over the course of 5 years (NIA, 2019). To achieve their mission, the TREAT-AD centers will “1) design, develop and disseminate tools that support target enabling packages (TEPs) for the experimental validation of novel, next generation therapeutic targets, including those emanating from the NIA-funded, target discovery programs such as Accelerating Medicines Partnership-Alzheimer’s Disease (AMP-AD), and 2) initiate early stage drug discovery campaigns against the enabled targets” (TREAT-AD, 2024). In doing so, the program aims to de-risk potential therapies to incentivize industry to continue investment in their development. The data and experimental and analytic tools generated from the work of TREAT-AD are available to investigators from industry, academia, and other research organizations. For example, the TEPs, which include “validated commercial antibodies, protein structures, genetically modified cell lines, and assay protocols,” and associated data can be accessed through the AD Knowledge Portal to aid researchers in validating novel and understudied targets (NIA, 2023c).
these conditions often overlap (as discussed earlier in this chapter). This overlap suggests a shared pathophysiology, which could potentially be targeted by similar therapeutic strategies. NIH-funded programs, such as AMP-AD, TREAT-AD, MODEL-AD, and CLEAR-AD, can be instrumental in this regard with the expansion of their mandate beyond AD and into related dementias. Where parallel programs for related dementias already exist (e.g., AMP-PDRD), opportunities for integration should be pursued to break down silos (discussed further in Chapter 5). This expansion and integration would not only enhance the understanding of shared disease mechanisms but also facilitate the development of novel therapeutic targets and personalized medicine approaches. By considering and prioritizing mechanisms that might have relevance across AD/ADRD and supporting
research to develop mechanism-specific translational biomarkers, these programs can help to diversify and validate potential targets, thereby increasing the likelihood of successful clinical trials. This comprehensive approach is crucial for advancing the fight against these debilitating conditions.
Conclusion 3-7: The expansion of NIH-funded early-discovery and development programs to accelerate and diversify the therapeutic pipeline beyond AD to include related dementias will enhance understanding of healthy aging and neurodegeneration and support the development of novel therapeutic targets and personalized medicine approaches relevant to shared disease mechanisms.
Conclusion 3-8: The NIH-funded drug discovery programs and infrastructure for diversifying the therapeutic pipeline need to be strengthened with long-term support to develop and deliver critical drug discovery input and translational biomarkers that reflect changes in pathways under investigation.
A well-recognized barrier to advancing interventions for AD/ADRD and many other diseases is the so-called valley of death—the large gap that spans early-stage scientific research (generally in academic settings) that provides mechanistic insights and identifies promising therapeutic targets and the later-stage, lower-risk programs that are more attractive to pharmaceutical companies and investors (Parrish et al., 2019). As a result of the valley of death, many potentially effective therapeutic strategies are not pursued. Strategies to bridge the valley of death can include
- establishing challenge programs to spur innovation and the acceleration of translational research;
- strategically targeting public or philanthropic funding to de-risk novel intervention approaches to incentivize private-sector investment and advance research on strategies that the pharmaceutical industry may not have an interest in, such as repurposing generic drugs (Smith, 2024); and
- creating public–private partnerships, which can draw on the talents of academia in discovery and those of industry in bringing therapies to market (Atkinson et al., 2023; HHS, 2012).
Partnerships also have a role in accelerating translational research outside of the drug development space, as discussed further in Chapter 5. Partnerships between different public entities (e.g., NIH partnership with housing or education departments) can facilitate more cross-disciplinary
approaches and better leverage existing investments that may affect brain health and AD/ADRD (Kremer, 2024).
RESEARCH PRIORITIES
The advancement of knowledge of risk and protective exposures for diverse populations, elucidation of the complex biological basis of AD/ADRD, and the rapid translation of basic and epidemiological research discoveries to clinical trials are essential to the development of effective strategies for the prevention and treatment of AD/ADRD. In recent years, NIH has made significant investments in basic research focused on elucidating diverse molecular and cellular processes of AD/ADRD vulnerability and resilience (NIA, 2023d, 2024f). Infrastructure investments to support these efforts have included the creation of large-scale, collaborative research programs to support the identification of disease mechanisms; application of emerging analytic approaches; development of novel therapeutic targets and open-science tools; and mechanisms to support early-stage clinical research. The committee identified four research priorities that build upon these existing efforts and investments. These research priorities are listed in Table 3-2 and include key scientific questions and near-term research opportunities that would advance progress on the research priorities.
| Research Priority | Key Scientific Questions | Near-Term Research Opportunities to Address Key Scientific Questions |
|---|---|---|
| 3-1: Identify factors driving AD/ADRD risk in diverse populations, particularly understudied and disproportionately affected groups, to better understand disease heterogeneity—including molecular subtypes and disparities in environmental exposures—and to identify prevention opportunities and advance health research equity. |
|
|
| Research Priority | Key Scientific Questions | Near-Term Research Opportunities to Address Key Scientific Questions |
|---|---|---|
| 3-2: Characterize the exposome and gene–environment interactions across the life course to gain insights into biological mechanisms and identify opportunities to reduce AD/ADRD risk and increase resilience. |
|
|
| Research Priority | Key Scientific Questions | Near-Term Research Opportunities to Address Key Scientific Questions |
|---|---|---|
| 3-3: Elucidate the genetic and other biological mechanisms underlying resilience and resistance to identify novel targets and effective strategies for AD/ADRD prevention and treatment. |
|
|
| Research Priority | Key Scientific Questions | Near-Term Research Opportunities to Address Key Scientific Questions |
|---|---|---|
| 3-4: Develop integrated molecular and cellular causal models to guide the identification of common mechanisms underlying AD/ADRD and their validation as novel targets for prevention and treatment. |
|
|
| Research Priority | Key Scientific Questions | Near-Term Research Opportunities to Address Key Scientific Questions |
|---|---|---|
|
|
REFERENCES
Abeliovich, A., and A. D. Gitler. 2016. Defects in trafficking bridge Parkinson’s disease pathology and genetics. Nature 539(7628):207-216.
ACTC-DS. 2024. Alzheimer’s clinical trials consortium—Down syndrome. https://www.actc-ds.org/ (accessed August 23, 2024).
AD Knowledge Portal. 2024a. M2OVE-AD: Molecular mechanisms of the vascular etiology of Alzheimer’s disease. https://adknowledgeportal.synapse.org/Explore/Programs/DetailsPage?Program=M2OVE-AD (accessed August 19, 2024).
AD Knowledge Portal. 2024b. Resilience-AD: Cognitive resilience to Alzheimer’s disease. https://adknowledgeportal.synapse.org/Explore/Programs/DetailsPage?Program=Resilience-AD (accessed October 22, 2024).
ADSP (Alzheimer’s Disease Sequencing Project). 2023. List of AD loci and genes with genetic evidence compiled by ADSP gene verification committee. https://adsp.niagads.org/gvc-top-hits-list/ (accessed August 23, 2024).
ADSP. 2024. ADSP overview. https://adsp.niagads.org/about/adsp-overview/ (accessed August 16, 2024).
Ahmed, C., H. J. Greve, C. Garza-Lombo, J. A. Malley, J. A. Johnson, Jr., A. L. Oblak, and M. L. Block. 2024. Peripheral HMGB1 is linked to O3 pathology of disease-associated astrocytes and amyloid. Alzheimer’s & Dementia 20(5):3551-3566.
Ahmed, R. M., M. Irish, J. van Eersel, A. Ittner, Y. D. Ke, A. Volkerling, J. van der Hoven, K. Tanaka, T. Karl, M. Kassiou, J. J. Kril, O. Piguet, J. Gotz, M. C. Kiernan, G. M. Halliday, J. R. Hodges, and L. M. Ittner. 2017. Mouse models of frontotemporal dementia: A comparison of phenotypes with clinical symptomatology. Neuroscience and Biobehavior Review 74(Pt A):126-138.
Aizenstein, H. J., R. D. Nebes, J. A. Saxton, J. C. Price, C. A. Mathis, N. D. Tsopelas, S. K. Ziolko, J. A. James, B. E. Snitz, P. R. Houck, W. Bi, A. D. Cohen, B. J. Lopresti, S. T. DeKosky, E. M. Halligan, and W. E. Klunk. 2008. Frequent amyloid deposition without significant cognitive impairment among the elderly. Archives of Neurology 65(11):1509-1517.
AlzForum. 2024. Research models. https://www.alzforum.org/research-models/alzheimers-disease (accessed August 19, 2024).
Amartumur, S., H. Nguyen, T. Huynh, T. S. Kim, R. S. Woo, E. Oh, K. K. Kim, L. P. Lee, and C. Heo. 2024. Neuropathogenesis-on-chips for neurodegenerative diseases. Nature Communications 15(1):2219.
AMP-PD. n.d. About AMP PD overview. https://amp-pd.org/about (accessed October 23, 2024).
An, Y., V. R. Varma, S. Varma, R. Casanova, E. Dammer, O. Pletnikova, C. W. Chia, J. M. Egan, L. Ferrucci, J. Troncoso, A. I. Levey, J. Lah, N. T. Seyfried, C. Legido-Quigley, R. O’Brien, and M. Thambisetty. 2018. Evidence for brain glucose dysregulation in Alzheimer’s disease. Alzheimer’s & Dementia 14(3):318-329.
Andrews, S. J., B. Fulton-Howard, and A. Goate. 2020. Interpretation of risk loci from genome-wide association studies of Alzheimer’s disease. Lancet Neurology 19(4):326-335.
Arafah, A., S. Khatoon, I. Rasool, A. Khan, M. A. Rather, K. A. Abujabal, Y. A. H. Faqih, H. Rashid, S. M. Rashid, S. Bilal Ahmad, A. Alexiou, and M. U. Rehman. 2023. The future of precision medicine in the cure of Alzheimer’s disease. Biomedicines 11(2):335.
Arboleda-Velasquez, J. F., F. Lopera, M. O’Hare, S. Delgado-Tirado, C. Marino, N. Chmielewska, K. L. Saez-Torres, D. Amarnani, A. P. Schultz, R. A. Sperling, D. Leyton-Cifuentes, K. Chen, A. Baena, D. Aguillon, S. Rios-Romenets, M. Giraldo, E. Guzmán-Vélez, D. J. Norton, E. Pardilla-Delgado, A. Artola, J. S. Sanchez, J. Acosta-Uribe, M. Lalli, K. S. Kosik, M. J. Huentelman, H. Zetterberg, K. Blennow, R. A. Reiman, J. Luo, Y. Chen, P. Thiyyagura, Y. Su, G. R. Jun, M. Naymik, X. Gai, M. Bootwalla, J. Ji, L. Shen, J. B. Miller, L. A. Kim, P. N. Tariot, K. A. Johnson, E. M. Reiman, Y. T. Quiroz. 2019. Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: A case report. Nature Medicine 25(11):1680-1683.
Arenaza-Urquijo, E. M., R. Boyle, K. Casaletto, K. J. Anstey, C. Vila-Castelar, A. Colverson, E. Palpatzis, J. M. Eissman, T. Kheng Siang Ng, S. Raghavan, M. Akinci, J. M. J. Vonk, L. S. Machado, P. P. Zanwar, H. L. Shrestha, M. Wagner, S. Tamburin, H. R. Sohrabi, S. Loi, D. Bartrés-Faz, D. B. Dubal, P. Vemuri, O. Okonkwo, T. J. Hohman, M. Ewers, R. F. Buckley, Reserve, Resilience and Protective Factors Professional Interest Area, Sex and Gender Professional Interest area and the ADDRESS! Special Interest Group. 2024. Sex and gender differences in cognitive resilience to aging and Alzheimer’s disease. Alzheimer’s & Dementia 20(8):5695-5719.
Argyriou, S., J. F. Fullard, J. M. Krivinko, D. Lee, T. S. Wingo, A. P. Wingo, R. A. Sweet, and P. Roussos. 2024. Beyond memory impairment: The complex phenotypic landscape of Alzheimer’s disease. Trends in Molecular Medicine 30(8):713-722.
Arun, P., F. Rossetti, D. M. Wilder, S. Sajja, S. A. Van Albert, Y. Wang, I. D. Gist, and J. B. Long. 2020. Blast exposure leads to accelerated cellular senescence in the rat brain. Frontiers in Neurology 11:438.
Asiamah, E. A., B. Feng, R. Guo, X. Yaxing, X. Du, X. Liu, J. Zhang, H. Cui, and J. Ma. 2024. The contributions of the endolysosomal compartment and autophagy to APOEε4 allele-mediated increase in Alzheimer’s disease risk. Journal of Alzheimer’s Disease 97(3):1007-1031.
Asken, B. M., P. A. Ljubenkov, A. M. Staffaroni, K. B. Casaletto, L. Vandevrede, Y. Cobigo, J. C. Rojas-Rodriguez, K. P. Rankin, J. Kornak, H. Heuer, J. Shigenaga, B. S. Appleby, A. C. Bozoki, K. Domoto-Reilly, N. Ghoshal, E. Huey, I. Litvan, J. C. Masdeu, M. F. Mendez, B. Pascual, P. Pressman, M. C. Tartaglia, W. Kremers, L. K. Forsberg, B. F. Boeve, A. L. Boxer, H. J. Rosen, J. H. Kramer, and ALLFTD Consortium Investigators. 2023. Plasma inflammation for predicting phenotypic conversion and clinical progression of autosomal dominant frontotemporal lobar degeneration. Journal of Neurology, Neurosurgery, and Psychiatry 94(7):541-549.
Atkinson, P. J., M. Swami, N. Ridgway, M. Roberts, J. Kinghorn, T. T. Warner, J. M. Staddon, and A. K. Takle. 2023. Advancing novel therapies for neurodegeneration through an innovative model for industry–academia collaborations: A decade of theEisai-UCL experience. Drug Discovery Today 28(10):103732.
Baker, M., I. R. Mackenzie, S. M. Pickering-Brown, J. Gass, R. Rademakers, C. Lindholm, J. Snowden, J. Adamson, A. D. Sadovnick, S. Rollinson, A. Cannon, E. Dwosh, D. Neary, S. Melquist, A. Richardson, D. Dickson, Z. Berger, J. Eriksen, T. Robinson, C. Zehr, C. A. Dickey, R. Crook, E. McGowan, D. Mann, B. Boeve, H. Feldman, and M. Hutton. 2006. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442(7105):916-919.
Bakulski, K., L. Rozek, D. Dolinoy, H. Paulson, and H. Hu. 2012. Alzheimer’s disease and environmental exposure to lead: The epidemiologic evidence and potential role of epigenetics. Current Alzheimer Research 9(5):563-573.
Balusu, S., R. Praschberger, E. Lauwers, B. De Strooper, and P. Verstreken. 2023. Neurodegeneration cell per cell. Neuron 111(6):767-786.
Banbury Center. 2024. Integrating exposomics into the biomedical enterprise. Summary Report of Conference, December 3-6, 2023. https://www.cshl.edu/wp-content/uploads/2024/03/Banbury_EXPOS_Output_Two-Pager-Final_20240205.pdf.
Bardehle, S., V. A. Rafalski, and K. Akassoglou. 2015. Breaking boundaries—coagulation and fibrinolysis at the neurovascular interface. Frontiers in Cellular Neuroscience 9:354.
Bartlett, B. J., P. Isakson, J. Lewerenz, H. Sanchez, R. W. Kotzebue, R. C. Cumming, G. L. Harris, I. P. Nezis, D. R. Schubert, A. Simonsen, and K. D. Finley. 2011. p62, Ref(2)P and ubiquitinated proteins are conserved markers of neuronal aging, aggregate formation and progressive autophagic defects. Autophagy 7(6):572-583.
Bateman, R. J., P. S. Aisen, B. De Strooper, N. C. Fox, C. A. Lemere, J. M. Ringman, S. Salloway, R. A. Sperling, M. Windisch, and C. Xiong. 2011. Autosomal-dominant Alzheimer’s disease: A review and proposal for the prevention of Alzheimer’s disease. Alzheimer’s Research & Therapy 3(1):1.
Bateman, R. J., C. Xiong, T. L. Benzinger, A. M. Fagan, A. Goate, N. C. Fox, D. S. Marcus, N. J. Cairns, X. Xie, T. M. Blazey, D. M. Holtzman, A. Santacruz, V. Buckles, A. Oliver, K. Moulder, P. S. Aisen, B. Ghetti, W. E. Klunk, E. McDade, R. N. Martins, C. L. Masters, R. Mayeux, J. M. Ringman, M. N. Rossor, P. R. Schofield, R. A. Sperling, S. Salloway, and J. C. Morris; Dominantly Inherited Alzheimer Network. 2012. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. New England Journal of Medicine 367(9):795-804.
Beckman, D., P. Chakrabarty, S. Ott, A. Dao, E. Zhou, W. G. Janssen, K. Donis-Cox, S. Muller, J. H. Kordower, and J. H. Morrison. 2021. A novel tau-based rhesus monkey model of Alzheimer’s pathogenesis. Alzheimer’s & Dementia 17(6):933-945.
Behl, C. 2023. Alzheimer’s disease research. Springer Cham.
Bellenguez, C., F. Kucukali, I. E. Jansen, L. Kleineidam, S. Moreno-Grau, N. Amin, A. C. Naj, R. Campos-Martin, B. Grenier-Boley, V. Andrade, P. A. Holmans, A. Boland, V. Damotte, S. J. van der Lee, M. R. Costa, T. Kuulasmaa, Q. Yang, I. de Rojas, J. C. Bis, A. Yaqub, I. Prokic, J. Chapuis, S. Ahmad, V. Giedraitis, D. Aarsland, P. Garcia-Gonzalez, C. Abdelnour, E. Alarcon-Martin, D. Alcolea, M. Alegret, I. Alvarez, V. Alvarez, N. J. Armstrong, A. Tsolaki, C. Antunez, I. Appollonio, M. Arcaro, S. Archetti, A. A. Pastor, B. Arosio, L. Athanasiu, H. Bailly, N. Banaj, M. Baquero, S. Barral, A. Beiser, A. B. Pastor, J. E. Below, P. Benchek, L. Benussi, C. Berr, C. Besse, V. Bessi, G. Binetti, A. Bizarro, R. Blesa, M. Boada, E. Boerwinkle, B. Borroni, S. Boschi, P. Bossu, G. Brathen, J. Bressler, C. Bresner, H. Brodaty, K. J. Brookes, L. I. Brusco, D. Buiza-Rueda, K. Burger, V. Burholt, W. S. Bush, M. Calero, L. B. Cantwell, G. Chene, J. Chung, M. L. Cuccaro, A. Carracedo, R. Cecchetti, L. Cervera-Carles, C. Charbonnier, H. H. Chen, C. Chillotti, S. Ciccone, J. Claassen, C. Clark, E. Conti, A. Corma-Gomez, E. Costantini, C. Custodero, D. Daian, M. C. Dalmasso, A. Daniele, E. Dardiotis, J. F. Dartigues, P. P. de Deyn, K. de Paiva Lopes, L. D. de Witte, S. Debette, J. Deckert, T. Del Ser, N. Denning, A. DeStefano, M. Dichgans, J. Diehl-Schmid, M. Diez-Fairen, P. D. Rossi, S. Djurovic, E. Duron, E. Duzel, C. Dufouil, G. Eiriksdottir, S. Engelborghs, V. Escott-Price, A. Espinosa, M. Ewers, K. M. Faber, T. Fabrizio, S. F. Nielsen, D. W. Fardo, L. Farotti, C. Fenoglio, M. Fernandez-Fuertes, R. Ferrari, C. B. Ferreira, E. Ferri, B. Fin, P. Fischer, T. Fladby, K. Fliessbach, B. Fongang, M. Fornage, J. Fortea, T. M. Foroud, S. Fostinelli, N. C. Fox, E. Franco-Macias, M. J. Bullido, A. Frank-Garcia, L. Froelich, B. Fulton-Howard, D. Galimberti, J. M. Garcia-Alberca, P. Garcia-Gonzalez, S. Garcia-Madrona, G. Garcia-Ribas, R. Ghidoni, I. Giegling, G. Giorgio, A. M. Goate, O. Goldhardt, D. Gomez-Fonseca, A. Gonzalez-Perez, C. Graff, G. Grande, E. Green, T. Grimmer, E. Grunblatt, M. Grunin, V. Gudnason, T. Guetta-Baranes, A. Haapasalo, G. Hadjigeorgiou, J. L. Haines, K. L. Hamilton-Nelson, H. Hampel, O. Hanon, J. Hardy, A. M. Hartmann, L. Hausner, J. Harwood, S. Heilmann-Heimbach, S. Helisalmi, M. T. Heneka, I. Hernandez, M. J. Herrmann, P. Hoffmann, C. Holmes, H. Holstege, R. H. Vilas, M. Hulsman, J. Humphrey, G. J. Biessels, X. Jian, C. Johansson, G. R. Jun, Y. Kastumata, J. Kauwe, P. G. Kehoe, L. Kilander, A. K. Stahlbom, M. Kivipelto, A. Koivisto, J. Kornhuber, M. H. Kosmidis, W. A. Kukull, P. P. Kuksa, B. W. Kunkle, A. B. Kuzma, C. Lage, E. J. Laukka, L. Launer, A. Lauria, C. Y. Lee, J. Lehtisalo, O. Lerch, A. Lleo, W. Longstreth, Jr., O. Lopez, A. L. de Munain, S. Love, M. Lowemark, L. Luckcuck, K. L. Lunetta, Y. Ma, J. Macias, C. A. MacLeod, W. Maier, F. Mangialasche, M. Spallazzi, M. Marquie, R. Marshall, E. R. Martin, A. M. Montes, C. M. Rodriguez, C. Masullo, R. Mayeux, S. Mead, P. Mecocci, M. Medina, A. Meggy, S. Mehrabian, S. Mendoza, M. Menendez-Gonzalez, P. Mir, S. Moebus, M. Mol, L. Molina-Porcel, L. Montrreal, L.
Morelli, F. Moreno, K. Morgan, T. Mosley, M. M. Nothen, C. Muchnik, S. Mukherjee, B. Nacmias, T. Ngandu, G. Nicolas, B. G. Nordestgaard, R. Olaso, A. Orellana, M. Orsini, G. Ortega, A. Padovani, C. Paolo, G. Papenberg, L. Parnetti, F. Pasquier, P. Pastor, G. Peloso, A. Perez-Cordon, J. Perez-Tur, P. Pericard, O. Peters, Y. A. L. Pijnenburg, J. A. Pineda, G. Pinol-Ripoll, C. Pisanu, T. Polak, J. Popp, D. Posthuma, J. Priller, R. Puerta, O. Quenez, I. Quintela, J. Q. Thomassen, A. Rabano, I. Rainero, F. Rajabli, I. Ramakers, L. M. Real, M. J. T. Reinders, C. Reitz, D. Reyes-Dumeyer, P. Ridge, S. Riedel-Heller, P. Riederer, N. Roberto, E. Rodriguez-Rodriguez, A. Rongve, I. R. Allende, M. Rosende-Roca, J. L. Royo, E. Rubino, D. Rujescu, M. E. Saez, P. Sakka, I. Saltvedt, A. Sanabria, M. B. Sanchez-Arjona, F. Sanchez-Garcia, P. S. Juan, R. Sanchez-Valle, S. B. Sando, C. Sarnowski, C. L. Satizabal, M. Scamosci, N. Scarmeas, E. Scarpini, P. Scheltens, N. Scherbaum, M. Scherer, M. Schmid, A. Schneider, J. M. Schott, G. Selbaek, D. Seripa, M. Serrano, J. Sha, A. A. Shadrin, O. Skrobot, S. Slifer, G. J. L. Snijders, H. Soininen, V. Solfrizzi, A. Solomon, Y. Song, S. Sorbi, O. Sotolongo-Grau, G. Spalletta, A. Spottke, A. Squassina, E. Stordal, J. P. Tartan, L. Tarraga, N. Tesi, A. Thalamuthu, T. Thomas, G. Tosto, L. Traykov, L. Tremolizzo, A. Tybjaerg-Hansen, A. Uitterlinden, A. Ullgren, I. Ulstein, S. Valero, O. Valladares, C. Van Broeckhoven, J. Vance, B. N. Vardarajan, A. van der Lugt, J. Van Dongen, J. van Rooij, J. van Swieten, R. Vandenberghe, F. Verhey, J. S. Vidal, J. Vogelgsang, M. Vyhnalek, M. Wagner, D. Wallon, L. S. Wang, R. Wang, L. Weinhold, J. Wiltfang, G. Windle, B. Woods, M. Yannakoulia, H. Zare, Y. Zhao, X. Zhang, C. Zhu, M. Zulaica, EADB, GR@ACE, DEGESCO, EADI, GERAD, Demgene, FinnGen, ADGC, CHARGE, L. A. Farrer, B. M. Psaty, M. Ghanbari, T. Raj, P. Sachdev, K. Mather, F. Jessen, M. A. Ikram, A. de Mendonca, J. Hort, M. Tsolaki, M. A. Pericak-Vance, P. Amouyel, J. Williams, R. Frikke-Schmidt, J. Clarimon, J. F. Deleuze, G. Rossi, S. Seshadri, O. A. Andreassen, M. Ingelsson, M. Hiltunen, K. Sleegers, G. D. Schellenberg, C. M. van Duijn, R. Sims, W. M. van der Flier, A. Ruiz, A. Ramirez, and J. C. Lambert. 2022. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nature Genetics 54(4):412-436.
Bhat, R., E. P. Crowe, A. Bitto, M. Moh, C. D. Katsetos, F. U. Garcia, F. B. Johnson, J. Q. Trojanowski, C. Sell, and C. Torres. 2012. Astrocyte senescence as a component of Alzheimer’s disease. PLoS ONE 7(9):e45069.
Bhattarai, P., T. Iniyan Gunasekaran, M. E. Belloy, D. Reyes-Dumeyer, D. Jülich, H. Tayran, E. Yilmaz, D. Flaherty, B. Turgutalp, G. Sukumar, C. Alba, E. Martinez McGrath, D. N. Hupalo, D. Bacikova, Y. Le Guen, R. Lantigua, M. Medrano, D. Rivera, P. Recio, T. Nuriel, N. Ertekin-Taner, A. F. Teich, D. W. Dickson, S. Holley, M. Greicius, C. L. Dalgard, M. Zody, R. Mayeux, C. Kizil, and B. N. Vardarajanet. 2024. Rare genetic variation in fibronectin 1 (FN1) protects against APOEε4 in Alzheimer’s disease. Acta Neuropathologica 147(1):70.
Biomarker Network. 2024. Health and Retirement Study (HRS). https://gero.usc.edu/cbph/network/studies-with-biomarkers/the-health-and-retirement-study-hrs/ (accessed August 16, 2024).
Bohnen, N. I., D. S. Djang, K. Herholz, Y. Anzai, and S. Minoshima. 2012. Effectiveness and safety of 18F-FDG PET in the evaluation of dementia: A review of the recent literature. Journal of Nuclear Medicine 53(1):59-71.
Bowles, K. R., M. C. Silva, K. Whitney, T. Bertucci, J. E. Berlind, J. D. Lai, J. C. Garza, N. C. Boles, S. Mahali, K. H. Strang, J. A. Marsh, C. Chen, D. A. Pugh, Y. Liu, R. E. Gordon, S. K. Goderie, R. Chowdhury, S. Lotz, K. Lane, J. F. Crary, S. J. Haggarty, C. M. Karch, J. K. Ichida, A. M. Goate, and S. Temple. 2021. ELAVL4, splicing, and glutamatergic dysfunction precede neuron loss in MAPT mutation cerebral organoids. Cell 184(17):4547-4563, e4517.
Bryant, A. G., M. Hu, B. C. Carlyle, S. E. Arnold, M. P. Frosch, S. Das, B. T. Hyman, and R. E. Bennett. 2020. Cerebrovascular senescence is associated with tau pathology in Alzheimer’s disease. Frontiers in Neurology 11:575953.
Bufill, E., R. Ribosa-Nogué, and R. Blesa. 2020. The therapeutic potential of epigenetic modifications in Alzheimer’s disease. In Alzheimer’s disease: Drug discovery. Edited by X. Huang. Brisbane, Australia: Exon Publications.
Bussian, T. J., A. Aziz, C. F. Meyer, B. L. Swenson, J. M. van Deursen, and D. J. Baker. 2018. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 562(7728):578-582.
Cacace, R., K. Sleegers, and C. Van Broeckhoven. 2016. Molecular genetics of early-onset Alzheimer’s disease revisited. Alzheimer‘s & Dementia 12(6):733-748.
Cai, W., K. Zhang, P. Li, L. Zhu, J. Xu, B. Yang, X. Hu, Z. Lu, and J. Chen. 2017. Dysfunction of the neurovascular unit in ischemic stroke and neurodegenerative diseases: An aging effect. Ageing Research Reviews 34:77-87.
Cao, W., and H. Zheng. 2018. Peripheral immune system in aging and Alzheimer’s disease. Molecular Neurodegeneration 13(1):51.
Carter, A. C., H. Y. Chang, G. Church, A. Dombkowski, J. R. Ecker, E. Gil, P. G. Giresi, H. Greely, W. J. Greenleaf, N. Hacohen, C. He, D. Hill, J. Ko, I. Kohane, A. Kundaje, M. Palmer, M. P. Snyder, J. Tung, U. Urban, M. Vidal, and W. Wong. 2017. Challenges and recommendations for epigenomics in precision health. Nature Biotechnology 35(12):1128-1132.
Cavaliere, F., and S. Gülöksüz. 2022. Shedding light on the etiology of neurodegenerative diseases and dementia: The exposome paradigm. NPJ Mental Health Research 1(1):20.
Cavalli, G., and E. Heard. 2019. Advances in epigenetics link genetics to the environment and disease. Nature 571(7766):489-499.
Cetin, S., D. Knez, S. Gobec, J. Kos and A. Pišlar. 2022. Cell models for Alzheimer’s and Parkinson’s disease: At the interface of biology and drug discovery. Biomedicine & Pharmacotherapy 149:112924.
Chakraborty, D., V. Felzen, C. Hiebel, E. Sturner, N. Perumal, C. Manicam, E. Sehn, F. Grus, U. Wolfrum, and C. Behl. 2019. Enhanced autophagic-lysosomal activity and increased BAG3-mediated selective macroautophagy as adaptive response of neuronal cells to chronic oxidative stress. Redox Biology 24:101181.
Chen, C., and J. M. Zissimopoulos. 2018. Racial and ethnic differences in trends in dementia prevalence and risk factors in the United States. Alzheimer’s & Dementia (N Y) 4:510-520.
Chen, X. and D. M. Holtzman. 2022. Emerging roles of innate and adaptive immunity in Alzheimer’s disease. Immunity 55(12):2236-2254.
Cheng, H., M. Wang, J. L. Li, N. J. Cairns, and X. Han. 2013. Specific changes of sulfatide levels in individuals with pre-clinical Alzheimer’s disease: An early event in disease pathogenesis. Journal of Neurochemistry 127(6):733-738.
Chia, R., M. S. Sabir, S. Bandres-Ciga, S. Saez-Atienzar, R. H. Reynolds, E. Gustavsson, R. L. Walton, S. Ahmed, C. Viollet, J. Ding, M. B. Makarious, M. Diez-Fairen, M. K. Portley, Z. Shah, Y. Abramzon, D. G. Hernandez, C. Blauwendraat, D. J. Stone, J. Eicher, L. Parkkinen, O. Ansorge, L. Clark, L. S. Honig, K. Marder, A. Lemstra, P. St George-Hyslop, E. Londos, K. Morgan, T. Lashley, T. T. Warner, Z. Jaunmuktane, D. Galasko, I. Santana, P. J. Tienari, L. Myllykangas, M. Oinas, N. J. Cairns, J. C. Morris, G. M. Halliday, V. M. Van Deerlin, J. Q. Trojanowski, M. Grassano, A. Calvo, G. Mora, A. Canosa, G. Floris, R. C. Bohannan, F. Brett, Z. Gan-Or, J. T. Geiger, A. Moore, P. May, R. Kruger, D. S. Goldstein, G. Lopez, N. Tayebi, E. Sidransky, American Genome Center, L. NorcliffeKaufmann, J. A. Palma, H. Kaufmann, V. G. Shakkottai, M. Perkins, K. L. Newell, T. Gasser, C. Schulte, F. Landi, E. Salvi, D. Cusi, E. Masliah, R. C. Kim, C. A. Caraway, E. S. Monuki, M. Brunetti, T. M. Dawson, L. S. Rosenthal, M. S. Albert, O. Pletnikova, J. C. Troncoso, M. E. Flanagan, Q. Mao, E. H. Bigio, E. Rodriguez-Rodriguez, J. Infante, C. Lage, I. Gonzalez-Aramburu, P. Sanchez-Juan, B. Ghetti, J. Keith, S. E. Black, M. Masellis, E. Rogaeva, C. Duyckaerts, A. Brice, S. Lesage, G. Xiromerisiou, M. J. Barrett, B. S. Tilley,
S. Gentleman, G. Logroscino, G. E. Serrano, T. G. Beach, I. G. McKeith, A. J. Thomas, J. Attems, C. M. Morris, L. Palmer, S. Love, C. Troakes, S. Al-Sarraj, A. K. Hodges, D. Aarsland, G. Klein, S. M. Kaiser, R. Woltjer, P. Pastor, L. M. Bekris, J. B. Leverenz, L. M. Besser, A. Kuzma, A. E. Renton, A. Goate, D. A. Bennett, C. R. Scherzer, H. R. Morris, R. Ferrari, D. Albani, S. Pickering-Brown, K. Faber, W. A. Kukull, E. Morenas-Rodriguez, A. Lleó, J. Fortea, D. Alcolea, J. Clarimon, M. A. Nalls, L. Ferrucci, S. M. Resnick, T. Tanaka, T. M. Foroud, N. R. Graff-Radford, Z. K. Wszolek, T. Ferman, B. F. Boeve, J. A. Hardy, E. J. Topol, A. Torkamani, A. B. Singleton, M. Ryten, D. W. Dickson, A. Chio, O. A. Ross, J. R. Gibbs, C. L. Dalgard, B. J. Traynor, and S. W. Scholz. 2021. Genome sequencing analysis identifies new loci associated with Lewy body dementia and provides insights into its genetic architecture. Nature Genetics 53(3):294-303.
Chow, H.-M., M. Shi, A. Cheng, Y. Gao, G. Chen, X. Song, R. W. L. So, J. Zhang, and K. Herrup. 2019. Age-related hyperinsulinemia leads to insulin resistance in neurons and cell-cycle-induced senescence. Nature Neuroscience 22(11):1806-1819.
Ciani, M., C. Bonvicini, C. Scassellati, M. Carrara, C. Maj, S. Fostinelli, G. Binetti, R. Ghidoni, and L. Benussi. 2019. The missing heritability of sporadic frontotemporal dementia: New insights from rare variants in neurodegenerative candidate genes. International Journal of Molecular Science 20(16):E3903.
Claassen, J., D. H. J. Thijssen, R. B. Panerai, and F. M. Faraci. 2021. Regulation of cerebral blood flow in humans: Physiology and clinical implications of autoregulation. Physiological Reviews 101(4):1487-1559.
Clark, K., Y. Y. Leung, W. P. Lee, B. Voight, and L. S. Wang. 2022. Polygenic risk scores in Alzheimer’s disease genetics: Methodology, applications, inclusion, and diversity. Journal of Alzheimer’s Disease 89(1):1-12.
Cocean, A. M., and D. C. Vodnar. 2024. Exploring the gut-brain axis: Potential therapeutic impact of psychobiotics on mental health. Progress in Neuro-Psychopharmacology & Biological Psychiatry 134:111073.
Collins, H. M., and S. Greenfield. 2024. Rodent models of Alzheimer’s disease: Past misconceptions and future prospects. International Journal of Molecular Science 25(11):6222.
Columbia University. 2024. NIH Award Creates Columbia-Led Exposomics Coordinating Center. https://www.publichealth.columbia.edu/news/nih-award-creates-columbia-led-exposomics-coordinating-center (accessed November 11, 2024).
Corriveau, R. A., F. Bosetti, M. Emr, J. T. Gladman, J. I. Koenig, C. S. Moy, K. Pahigiannis, S. P. Waddy, and W. Koroshetz. 2016. The science of vascular contributions to cognitive impairment and dementia (VCID): A framework for advancing research priorities in the cerebrovascular biology of cognitive decline. Cellular and Molecular Neurobiology 36(2):281-288.
Crimmins, E. M. 2020. Social hallmarks of aging: Suggestions for geroscience research. Ageing Research Review 63:101136.
Cruts, M., I. Gijselinck, J. van der Zee, S. Engelborghs, H. Wils, D. Pirici, R. Rademakers, R. Vandenberghe, B. Dermaut, J.-J. Martin, C. van Duijn, K. Peeters, R. Sciot, P. Santens, T. De Pooter, M. Mattheijssens, M. Van den Broeck, I. Cuijt, K. l. Vennekens, P. P. De Deyn, S. Kumar-Singh, and C. Van Broeckhoven. 2006. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature 442(7105):920-924.
Cummings, J., A. M. Leisgang Osse, and J. Kinney. 2023a. Geroscience and Alzheimer’s disease drug development. Journal of Prevention of Alzheimer’s Disease 10(4):620-632.
Cummings, J., Y. Zhou, G. Lee, K. Zhong, J. Fonseca, and F. Cheng. 2023b. Alzheimer’s disease drug development pipeline: 2023. Alzheimer’s & Dementia (N Y) 9(2):e12385.
Cutler, R. G., J. Kelly, K. Storie, W. A. Pedersen, A. Tammara, K. Hatanpaa, J. C. Troncoso, and M. P. Mattson. 2004. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proceedings of the National Academy of Sciences USA 101(7):2070-2075.
de Galan, B. E. 2024. Diabetes and brain disorders, a new role for insulin? Neuroscience and Biobehavioral Review 163:105775.
De Jager, P. L., G. Srivastava, K. Lunnon, J. Burgess, L. C. Schalkwyk, L. Yu, M. L. Eaton, B. T. Keenan, J. Ernst, C. McCabe, A. Tang, T. Raj, J. Replogle, W. Brodeur, S. Gabriel, H. S. Chai, C. Younkin, S. G. Younkin, F. Zou, M. Szyf, C. B. Epstein, J. A. Schneider, B. E. Bernstein, A. Meissner, N. Ertekin-Taner, L. B. Chibnik, M. Kellis, J. Mill, and D. A. Bennett. 2014. Alzheimer’s disease: Early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nature Neuroscience 17(9):1156-1163.
de la Monte, S. M., T. Luong, T. R. Neely, D. Robinson, and J. R. Wands. 2000. Mitochondrial DNA damage as a mechanism of cell loss in Alzheimer’s disease. Laboratory Investigation 80(8):1323-1335.
De Strooper, B., and E. Karran. 2016. The cellular phase of Alzheimer’s disease. Cell 164(4):603-615.
de Vries, L. E., I. Huitinga, H. W. Kessels, D. F. Swaab, and J. Verhaagen. 2024. The concept of resilience to Alzheimer’s disease: Current definitions and cellular and molecular mechanisms. Molecular Neurodegeneration 19(1):33.
Dehkordi, S. K., J. Walker, E. Sah, E. Bennett, F. Atrian, B. Frost, B. Woost, R. E. Bennett, T. C. Orr, Y. Zhou, P. S. Andhey, M. Colonna, P. H. Sudmant, P. Xu, M. Wang, B. Zhang, H. Zare, and M. E. Orr. 2021. Profiling senescent cells in human brains reveals neurons with CDKN2D/p19 and tau neuropathology. Nature Aging 1(12):1107-1116.
Deng, Z., P. Sheehan, S. Chen, and Z. Yue. 2017. Is amyotrophic lateral sclerosis/frontotemporal dementia an autophagy disease? Molecular Neurodegeneration 12(1):90.
Drzezga, A., N. Lautenschlager, H. Siebner, M. Riemenschneider, F. Willoch, S. Minoshima, M. Schwaiger, and A. Kurz. 2003. Cerebral metabolic changes accompanying conversion of mild cognitive impairment into Alzheimer’s disease: A PET follow-up study. European Journal of Nuclear Medicine and Molecular Imaging 30(8):1104-1113.
Du, Y., W. Deng, Z. Wang, M. Ning, W. Zhang, Y. Zhou, E. H. Lo, and C. Xing. 2017. Differential subnetwork of chemokines/cytokines in human, mouse, and rat brain cells after oxygen-glucose deprivation. Journal of Cerebral Blood Flow and Metabolism 37(4):1425-1434.
Duelli, R., and W. Kuschinsky. 2001. Brain glucose transporters: Relationship to local energy demand. News in Physiological Sciences 16:71-76.
Duffy, S. S., B. A. Keating, C. J. Perera, and G. Moalem-Taylor. 2018. The role of regulatory T-cells in nervous system pathologies. Journal of Neuroscience Research 96(6):951-968.
Dunn, A. R., K. M. S. O’Connell, and C. C. Kaczorowski. 2019. Gene-by-environment interactions in Alzheimer’s disease and Parkinson’s disease. Neuroscience & Biobehavioral Reviews 103:73-80.
Durso, D. F., G. Silveira-Nunes, M. M. Coelho, G. C. Camatta, L. H. Ventura, L. S. Nascimento, F. Caixeta, E. H. M. Cunha, A. Castelo-Branco, D. M. Fonseca, T. U. Maioli, A. Teixeira-Carvalho, C. Sala, M. J. Bacalini, P. Garagnani, C. Nardini, C. Franceschi, and A. M. C. Faria. 2022. Living in endemic area for infectious diseases accelerates epigenetic age. Mechanisms of Ageing and Development 207:111713.
Dutta, M., K. M. Weigel, K. T. Patten, A. E. Valenzuela, C. Wallis, K. J. Bein, A. S. Wexler, P. J. Lein, and J. Y. Cui. 2022. Chronic exposure to ambient traffic-related air pollution (TRAP) alters gut microbial abundance and bile acid metabolism in a transgenic rat model of Alzheimer’s disease. Toxicology Report 9:432-444.
Favier, A., G. Rocher, A. K. Larsen, R. Delangle, C. Uzan, M. Sabbah, M. Castela, A. Duval, C. Mehats, and G. Canlorbe. 2021. MicroRNA as epigenetic modifiers in endometrial cancer: A systematic review. Cancers (Basel) 13(5):1137.
Ferrari, R., D. G. Hernandez, M. A. Nalls, J. D. Rohrer, A. Ramasamy, J. B. Kwok, C. Dobson-Stone, W. S. Brooks, P. R. Schofield, G. M. Halliday, J. R. Hodges, O. Piguet, L. Bartley, E. Thompson, E. Haan, I. Hernandez, A. Ruiz, M. Boada, B. Borroni, A. Padovani, C. Cruchaga, N. J. Cairns, L. Benussi, G. Binetti, R. Ghidoni, G. Forloni, D. Galimberti, C. Fenoglio, M. Serpente, E. Scarpini, J. Clarimon, A. Lleo, R. Blesa, M. L. Waldo, K. Nilsson, C. Nilsson, I. R. Mackenzie, G. Y. Hsiung, D. M. Mann, J. Grafman, C. M. Morris, J. Attems, T. D. Griffiths, I. G. McKeith, A. J. Thomas, P. Pietrini, E. D. Huey, E. M. Wassermann, A. Baborie, E. Jaros, M. C. Tierney, P. Pastor, C. Razquin, S. Ortega-Cubero, E. Alonso, R. Perneczky, J. Diehl-Schmid, P. Alexopoulos, A. Kurz, I. Rainero, E. Rubino, L. Pinessi, E. Rogaeva, P. St George-Hyslop, G. Rossi, F. Tagliavini, G. Giaccone, J. B. Rowe, J. C. Schlachetzki, J. Uphill, J. Collinge, S. Mead, A. Danek, V. M. Van Deerlin, M. Grossman, J. Q. Trojanowski, J. van der Zee, W. Deschamps, T. Van Langenhove, M. Cruts, C. Van Broeckhoven, S. F. Cappa, I. Le Ber, D. Hannequin, V. Golfier, M. Vercelletto, A. Brice, B. Nacmias, S. Sorbi, S. Bagnoli, I. Piaceri, J. E. Nielsen, L. E. Hjermind, M. Riemenschneider, M. Mayhaus, B. Ibach, G. Gasparoni, S. Pichler, W. Gu, M. N. Rossor, N. C. Fox, J. D. Warren, M. G. Spillantini, H. R. Morris, P. Rizzu, P. Heutink, J. S. Snowden, S. Rollinson, A. Richardson, A. Gerhard, A. C. Bruni, R. Maletta, F. Frangipane, C. Cupidi, L. Bernardi, M. Anfossi, M. Gallo, M. E. Conidi, N. Smirne, R. Rademakers, M. Baker, D. W. Dickson, N. R. Graff-Radford, R. C. Petersen, D. Knopman, K. A. Josephs, B. F. Boeve, J. E. Parisi, W. W. Seeley, B. L. Miller, A. M. Karydas, H. Rosen, J. C. van Swieten, E. G. Dopper, H. Seelaar, Y. A. Pijnenburg, P. Scheltens, G. Logroscino, R. Capozzo, V. Novelli, A. A. Puca, M. Franceschi, A. Postiglione, G. Milan, P. Sorrentino, M. Kristiansen, H. H. Chiang, C. Graff, F. Pasquier, A. Rollin, V. Deramecourt, F. Lebert, D. Kapogiannis, L. Ferrucci, S. Pickering-Brown, A. B. Singleton, J. Hardy, and P. Momeni. 2014. Frontotemporal dementia and its subtypes: A genome-wide association study. Lancet Neurology 13(7):686-699.
Fillit, H. M., B. Vellas, and Y. Hara. 2023. Editorial: The state of Alzheimer’s research and the path forward. Journal of the Prevention of Alzheimer’s Disease 10(4):617-619.
Finch, C. E., and A. M. Kulminski. 2019. The Alzheimer’s disease exposome. Alzheimer’s & Dementia 15(9):1123-1132.
FNIH (Foundation for the National Institutes of Health). 2024. AMP Parkinson’s Disease and related disorders. https://fnih.org/our-programs/accelerating-medicines-partnership-amp/parkinsons-disease-and-related-disorders/ (accessed October 23, 2024).
Fortea, J., S. H. Zaman, S. Hartley, M. S. Rafii, E. Head, and M. Carmona-Iragui. 2021. Alzheimer’s disease associated with Down syndrome: A genetic form of dementia. Lancet Neurology 20(11):930-942.
Fortea, J., J. Pegueroles, D. Alcolea, O. Belbin, O. Dols-Icardo, L. Vaqué-Alcázar, L. Videla, J. Domingo Gispert, M. Suárez-Calvet, S. C. Johnson, R. Sperling, A. Bejanin, A. Lleó, and V. Montal. 2024. APOE4 homozygosity represents a distinct genetic form of Alzheimer’s disease. Nature Medicine 30(5):1284-1291.
Franceschi, C., and J. Campisi. 2014. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. Journals of Gerontology: Series A, Biological Sciences and Medical Sciences 69(Suppl 1):S4-S9.
Gan, L., M. R. Cookson, L. Petrucelli, and A. R. La Spada. 2018. Converging pathways in neurodegeneration, from genetics to mechanisms. Nature Neuroscience 21(10):1300-1309.
Gao, C., J. Jiang, Y. Tan, and S. Chen. 2023. Microglia in neurodegenerative diseases: Mechanism and potential therapeutic targets. Signal Transduction and Targeted Therapy 8: 359.
Gao, X., Q. Chen, H. Yao, J. Tan, Z. Liu, Y. Zhou, and Z. Zou. 2022. Epigenetics in Alzheimer’s disease. Frontiers in Aging and Neuroscience 14:911635.
Gate, D., E. Tapp, O. Leventhal, M. Shahid, T. J. Nonninger, A. C. Yang, K. Strmpfl, M. Unger, T. Fehlmann, H. Oh, D. Channappa, V. Henderson, A. Keller, L. Aigner, G. Galasko, M. Davis, K. Poston, and T. Wyss-Coray. 2021. CD4+ T cells contribute to neurodegeneration in Lewy body dementia. Science 374(6569):868-874.
Gatz, M., C. Reynolds, L. Fratiglioni, B. Johansson, J. Mortimer, S. Berg, A. Fiske, and N. Petdersen. 2006. Role of genes and environments for explaining Alzheimer’s disease. Archives of General Psychiatry 63(2):168-174.
Gendelman, H. E., and S. H. Appel. 2011. Neuroprotective activities of regulatory T cells. Trends in Molecular Medicine 17(12):687-688.
Genin, E., D. Hannequin, D. Wallon, K. Sleegers, M. Hiltunen, O. Combarros, M. J. Bullido, S. Engelborghs, P. De Deyn, C. Berr, F. Pasquier, B. Dubois, G. Tognoni, N. Fiévet, N. Brouwers, K. Bettens, B. Arosio, E. Coto, M. Del Zompo, I. Mateo, J. Epelbaum, A. Frank-Garcia, S. Helisalmi, E. Porcellini, A. Pilotto, P. Forti, R. Ferri, E. Scarpini, G. Siciliano, V. Solfrizzi, S. Sorbi, G. Spalletta, F. Valdivieso, S. Vepsäläinen, V. Alvarez, P. Bosco, M. Mancuso, F. Panza, B. Nacmias, P. Bossù, O. Hanon, P. Piccardi, G. Annoni, D. Seripa, D. Galimberti, F. Licastro, H. Soininen, J. F. Dartigues, M. I. Kamboh, C. Van Broeckhoven, J. C. Lambert, P. Amouyel, and D. Campion D. 2011. APOE and Alzheimer disease: A major gene with semi-dominant inheritance. Molecular Psychiatry 16(9):903-907.
Gifre-Renom, L., M. Daems, A. Luttun, and E. A. V. Jones. 2022. Organ-specific endothelial cell differentiation and impact of microenvironmental cues on endothelial heterogeneity. International Journal of Molecular Science 23(3):1477.
Golpich, M., E. Amini, Z. Mohamed, R. Azman Ali, N. Mohamed Ibrahim, and A. Ahmadiani. 2017. Mitochondrial dysfunction and biogenesis in neurodegenerative diseases: Pathogenesis and treatment. CNS Neuroscience and Therapy 23(1):5-22.
Gonzales, M. M., V. R. Garbarino, E. Pollet, J. P. Palavicini, D. L. Kellogg, Jr., E. Kraig, and M. E. Orr. 2022. Biological aging processes underlying cognitive decline and neurodegenerative disease. Journal of Clinical Investigation 132(10):e158453.
Gonzalez, H. M., W. Tarraf, A. M. Stickel, A. Morlett, K. A. Gonzalez, A. R. Ramos, T. Rundek, L. C. Gallo, G. A. Talavera, M. L. Daviglus, R. B. Lipton, C. Isasi, M. Lamar, D. Zeng, and C. DeCarli. 2024. Glycemic control, cognitive aging, and impairment among diverse Hispanic/Latino individuals: Study of Latinos—Investigation of neurocognitive aging (Hispanic Community Health Study/Study of Latinos). Diabetes Care 47(7):1152-1161.
Goodenowe, D. B., L. L. Cook, J. Liu, Y. Lu, D. A. Jayasinghe, P. W. Ahiahonu, D. Heath, Y. Yamazaki, J. Flax, K. F. Krenitsky, D. L. Sparks, A. Lerner, R. P. Friedland, T. Kudo, K. Kamino, T. Morihara, M. Takeda, and P. L. Wood. 2007. Peripheral ethanolamine plasmalogen deficiency: A logical causative factor in Alzheimer’s disease and dementia. Journal of Lipid Research 48(11):2485-2498.
Gorelick, P. B., A. Scuteri, S. E. Black, C. Decarli, S. M. Greenberg, C. Iadecola, L. J. Launer, S. Laurent, O. L. Lopez, D. Nyenhuis, R. C. Petersen, J. A. Schneider, C. Tzourio, D. K. Arnett, D. A. Bennett, H. C. Chui, R. T. Higashida, R. Lindquist, P. M. Nilsson, G. C. Roman, F. W. Sellke, S. Seshadri, American Heart Association Stroke Council, Council on Epidemiology and Prevention, Council on Cardiovascular Nursing, Council on Cardiovascular Radiology and Intervention, and Council on Cardiovascular Surgery and Anesthesia. 2011. Vascular contributions to cognitive impairment and dementia: A statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 42(9):2672-2713.
Gorgoulis, V., P. D. Adams, A. Alimonti, D. C. Bennett, O. Bischof, C. Bishop, J. Campisi, M. Collado, K. Evangelou, G. Ferbeyre, J. Gil, E. Hara, V. Krizhanovsky, D. Jurk, A. B. Maier, M. Narita, L. Niedernhofer, J. F. Passos, P. D. Robbins, C. A. Schmitt, J. Sedivy, K. Vougas, T. von Zglinicki, D. Zhou, M. Serrano, and M. Demaria. 2019. Cellular senescence: Defining a path forward. Cell 179(4):813-827.
Greaves, C. V., and J. D. Rohrer. 2019. An update on genetic frontotemporal dementia. Journal of Neurology 266(8):2075-2086.
Greve, H. J., A. L. Dunbar, C. G. Lombo, C. Ahmed, M. Thang, E. J. Messenger, C. L. Mumaw, J. A. Johnson, U. P. Kodavanti, A. L. Oblak, and M. L. Block. 2023. The bidirectional lung brain-axis of amyloid-beta pathology: Ozone dysregulates the peri-plaque microenvironment. Brain 146(3):991-1005.
Grodstein, F., B. Lemos, L. Yu, A. Iatrou, P. L. De Jager, and D. A. Bennett. 2020. Characteristics of epigenetic clocks across blood and brain tissue in older women and men. Frontiers in Neuroscience 14:555307.
Guan, P. P., and P. Wang. 2023. The involvement of post-translational modifications in regulating the development and progression of Alzheimer’s disease. Molecular Neurobiology 60(7):3617-3632.
Guerreiro, R., A. Wojtas, J. Bras, M. Carrasquillo, E. Rogaeva, E. Majounie, C. Cruchaga, C. Sassi, J. S. Kauwe, S. Younkin, L. Hazrati, J. Collinge, J. Pocock, T. Lashley, J. Williams, J. C. Lambert, P. Amouyel, A. Goate, R. Rademakers, K. Morgan, J. Powell, P. St George-Hyslop, A. Singleton, J. Hardy, and the Alzheimer Genetic Analysis Group. 2013. TREM2 variants in Alzheimer’s disease. New England Journal of Medicine 368(2):117-127.
Guerreiro, R., E. Gibbons, M. Tabuas-Pereira, C. Kun-Rodrigues, G. C. Santo, and J. Bras. 2020. Genetic architecture of common non-Alzheimer’s disease dementias. Neurobiology of Disease 142:104946.
Guo, P., W. Gong, Y. Li, L. Liu, R. Yan, Y. Wang, Y. Zhang, and Z. Yuan. 2022. Pinpointing novel risk loci for Lewy body dementia and the shared genetic etiology with Alzheimer’s disease and Parkinson’s disease: A large-scale multi-trait association analysis. BMC Medicine 20(1):214.
Hamamoto, R., M. Komatsu, K. Takasawa, K. Asada, and S. Kaneko. 2020. Epigenetics analysis and integrated analysis of multiomics data, including epigenetic data, using artificial intelligence in the era of precision medicine. Biomolecules 10(1):62.
Hampel, H., A. Vergallo, G. Perry, S. Lista, and the Alzheimer Precision Medicine Initiative. 2019. The Alzheimer’s Precision Medicine Initiative. Journal of Alzheimer’s Disease 68(1):1-24.
Hampel, H., F. Caraci, A. C. Cuello, G. Caruso, R. Nistico, M. Corbo, F. Baldacci, N. Toschi, F. Garaci, P. A. Chiesa, S. R. Verdooner, L. Akman-Anderson, F. Hernandez, J. Avila, E. Emanuele, P. L. Valenzuela, A. Lucia, M. Watling, B. P. Imbimbo, A. Vergallo, and S. Lista. 2020. A path toward precision medicine for neuroinflammatory mechanisms in Alzheimer’s disease. Frontiers in Immunology 11:456.
Han, X. 2010. Multi-dimensional mass spectrometry-based shotgun lipidomics and the altered lipids at the mild cognitive impairment stage of Alzheimer’s disease. Biochimica et Biophysica Acta 1801(8):774-783.
Han, X., A. M. Fagan, H. Cheng, J. C. Morris, C. Xiong, and D. M. Holtzman. 2003. Cerebrospinal fluid sulfatide is decreased in subjects with incipient dementia. Annals of Neurology 54(1):115-119.
Han, X., D. M. Holtzman, D. W. McKeel, Jr., J. Kelley, and J. C. Morris. 2002. Substantial sulfatide deficiency and ceramide elevation in very early Alzheimer’s disease: Potential role in disease pathogenesis. Journal of Neurochemistry 82(4):809-818.
Hara, Y., N. McKeehan, and H. M. Fillit. 2019. Translating the biology of aging into novel therapeutics for Alzheimer disease. Neurology 92(2):84-93.
Hargrove-Grimes, P., L. A. Low, and D. A. Tagle. 2021. Microphysiological systems: What it takes for community adoption. Experimental Biology and Medicine (Maywood) 246(12):1435-1446.
Harman, D. 1956. Aging: A theory based on free radical and radiation chemistry. Journal of Gerontology 11(3):298-300.
Harrison, J. R., S. Mistry, N. Muskett, and V. Escott-Price. 2020. From polygenic scores to precision medicine in Alzheimer’s disease: A systematic review. Journal of Alzheimer’s Disease 74:1271-1283.
Hernán, M. A. 2018. The C-word: Scientific euphemisms do not improve causal inference from observational data. American Journal of Public Health 108(5):616-619.
HHS (U.S. Department of Health and Human Services). 2012. Making the case for public private partnerships for NAPA. https://aspe.hhs.gov/sites/default/files/aspe-files/cmtach121.pdf (accessed October 18, 2024).
Ho, K. M., D. J. Morgan, M. Johnstone, and C. Edibam. 2023. Biological age is superior to chronological age in predicting hospital mortality of the critically ill. Internal Emergency Medicine 18(7):2019-2028.
Hooten, N., N. L. Pacheco, J. T. Smith, and M. K. Evans. 2022. The accelerated aging phenotype: The role of race and social determinants of health on aging. Ageing Research Review 73:101536.
Horvath, S., and A. J. Levine. 2015. HIV-1 infection accelerates age according to the epigenetic clock. Journal of Infectious Diseases 212(10):1563-1573.
Hu, Y. B., E. B. Dammer, R. J. Ren, and G. Wang. 2015. The endosomal-lysosomal system: From acidification and cargo sorting to neurodegeneration. Translational Neurodegeneration 4:18.
Huang, A. R., D. L. Roth, T. Cidav, S. E. Chung, H. Amjad, R. J. Thorpe, Jr., C. M. Boyd, and T. K. M. Cudjoe. 2023. Social isolation and 9-year dementia risk in community-dwelling Medicare beneficiaries in the United States. Journal of the American Geriatric Society 71(3):765-773.
Huq, A. J., P. Fransquet, S. M. Laws, J. Ryan, R. Sebra, C. L. Masters, I. M. Winship, P. A. James, and P. Lacaze. 2019. Genetic resilience to Alzheimer’s disease in APOE ε4 homozygotes: A systematic review. Alzheimer’s & Dementia 15(12):1612-1623.
Hurst, C., D. A. Pugh, M. H. Abreha, D. M. Duong, E. B. Dammer, D. A. Bennett, J. H. Herskowitz, and N. T. Seyfried. 2023. Integrated proteomics to understand the role of neuritin (NRN1) as a mediator of cognitive resilience to Alzheimer’s disease. Molecular & Cellular Proteomics 22(5):100542.
Ikram, M. A., A. Bersano, R. Manso-Calderón, J.-P. Jia, H. Schmidt, L. Middleton, B. Nacmias, S. Siddiqi, and H. H. H. Adams. 2017. Genetics of vascular dementia—review from the ICVD working group. BMC Medicine 15(1):48.
Irizarry, M. C. 2003. A turn of the sulfatide in Alzheimer’s disease. Annals of Neurology 54(1):7-8.
Italiani, P., I. Puxeddu, S. Napoletano, E. Scala, D. Melillo, S. Manocchio, A. Angiolillo, P. Migliorini, D. Boraschi, E. Vitale, and A. Di Costanzo. 2018. Circulating levels of IL-1 family cytokines and receptors in Alzheimer’s disease: New markers of disease progression? Journal of Neuroinflammation 15(1):342.
Itzhaki, R. F. 2021. Overwhelming evidence for a major role for herpes simplex virus type 1 (HSV1) in Alzheimer’s disease (AD); underwhelming evidence against. Vaccines (Basel) 9(6):679.
Iwata, A., K. Nagata, H. Hatsuta, H. Takuma, M. Bundo, K. Iwamoto, A. Tamaoka, S. Murayama, T. Saido, and S. Tsuji. 2014. Altered CpG methylation in sporadic Alzheimer’s disease is associated with APP and MAPT dysregulation. Human Molecular Genetics 23(3):648-656.
Jabbari, E., J. Woodside, M. M. X. Tan, M. Shoai, A. Pittman, R. Ferrari, K. Y. Mok, D. Zhang, R. H. Reynolds, R. de Silva, M. J. Grimm, G. Respondek, U. Muller, S. Al-Sarraj, S. M. Gentleman, A. J. Lees, T. T. Warner, J. Hardy, T. Revesz, G. U. Hoglinger, J. L. Holton, M. Ryten, and H. R. Morris. 2018. Variation at the TRIM11 locus modifies progressive supranuclear palsy phenotype. Annals of Neurology 84(4):485-496.
Janbek, J., T. M. Laursen, N. Frimodt-Møller, M. Magyari, J. G. Haas, R. Lathe, and G. Waldemar. 2023. Hospital-diagnosed infections, autoimmune diseases, and subsequent dementia incidence. JAMA Network Open 6(9):e2332635.
Jayaraj, R. L., E. A. Rodriguez, Y. Wang, and M. L. Block. 2017. Outdoor ambient air pollution and neurodegenerative diseases: The neuroinflammation hypothesis. Current Environmental Health Reports 4(2):166-179.
Jazwinski, S. M., and S. Kim. 2019. Examination of the dimensions of biological age. Frontiers in Genetics 10:263.
Jellinger, K. A. 2022. Recent update on the heterogeneity of the Alzheimer’s disease spectrum. Journal of Neural Transmission (Vienna) 129(1):1-24.
Jellinger, K. A., and A. D. Korczyn. 2018. Are dementia with Lewy bodies and Parkinson’s disease dementia the same disease? BMC Medicine 16(1):34.
Jia, L., F. Li, C. Wei, M. Zhu, Q. Qu, W. Qin, Y. Tang, L. Shen, Y. Wang, L. Shen, H. Li, D. Peng, L. Tan, B. Luo, Q. Guo, M. Tang, Y. Du, J. Zhang, J. Zhang, J. Lyu, Y. Li, A. Zhou, F. Wang, C. Chu, H. Song, L. Wu, X. Zuo, Y. Han, J. Liang, Q. Wang, H. Jin, W. Wang, Y. Lü, F. Li, Y. Zhou, W. Zhang, Z. Liao, Q. Qiu, Y. Li, C. Kong, Y. Li, H. Jiao, J. Lu, and J. Jia. 2021. Prediction of Alzheimer’s disease using multi-variants from a chinese genome-wide association study. Brain 144(3):924-937.
Jones, L., D. Harold, and J. Williams. 2010. Genetic evidence for the involvement of lipid metabolism in Alzheimer’s disease. Biochimica et Biophysica Acta 1801(8):754-761.
Jonsson, T., H. Stefansson, S. Steinberg, I. Jonsdottir, P. V. Jonsson, J. Snaedal, S. Bjornsson, J. Huttenlocher, A. I. Levey, J. J. Lah, D. Rujescu, H. Hampel, I. Giegling, O. A. Andreassen, K. Engedal, I. Ulstein, S. Djurovic, C. Ibrahim-Verbaas, A. Hofman, M. A. Ikram, C. M. van Duijn, U. Thorsteinsdottir, A. Kong, and K. Stefansson. 2013. Variant of TREM2 associated with the risk of Alzheimer’s disease. New England Journal of Medicine 368(2):107-116.
Kalia, V., D. W. Belsky, A. A. Baccarelli, and G. W. Miller. 2022. An exposomic framework to uncover environmental drivers of aging. Exposome 2(1):osac002.
Kapasi, A., C. DeCarli, and J. A. Schneider. 2017. Impact of multiple pathologies on the threshold for clinically overt dementia. Acta Neuropathologica 134(2):171-186.
Kashyap, S. N., N. R. Boyle, and E. D. Roberson. 2023. Preclinical interventions in mouse models of frontotemporal dementia due to progranulin mutations. Neurotherapeutics 20(1):140-153.
Kato, T., Y. Inui, A. Nakamura, and K. Ito. 2016. Brain fluorodeoxyglucose (FDG) PET in dementia. Ageing Research Reviews 30:73-84.
Katzman, R., R. Terry, R. DeTeresa, T. Brown, P. Davies, P. Fuld, X. Renbing, and A. Peck. 1988. Clinical, pathological, and neurochemical changes in dementia: A subgroup with preserved mental status and numerous neocortical plaques. Annals of Neurology 23(2):138-144.
Kelley, M. 2023. NIH strategic planning process overview. Presentation at Committee on Research Priorities for Preventing and Treating Alzheimer’s Disease and Related Dementias. Meeting 1, Virtual. October 2, 2024.
Kim, S. H., K. W. Oh, H. K. Jin, and J. S. Bae. 2018. Immune inflammatory modulation as a potential therapeutic strategy of stem cell therapy for ALS and neurodegenerative diseases. BMB Reports 51(11):545-546.
Kim, K., X. Wang, E. Ragonnaud, M. Bodogai, T. Illouz, M. DeLuca, R. A. McDevitt, F. Gusev, E. Okun, E. Rogaev, and A. Biragyn. 2021. Therapeutic B-cell depletion reverses progression of Alzheimer’s disease. Nature Communications 12:2185.
Kind, A. J. 2023. Establishing national social exposome Alzheimer’s disease and related dementias (ADRD) infrastructure. Alzheimer’s & Dementia 19(S22): e077828.
Kind, I. 2024. Panelist Remarks. Committee on Research Priorities for Preventing and Treating Alzheimer’s Disease and Related Dementias Public Workshop, Hybrid. January 16–17, 2024.
Klocke, C., and P. Lein. 2019. Environmental exposures during early life influence adult disease risk. https://www.openaccessgovernment.org/environmental-exposures-adult-disease-risk/75696/ (accessed August 16, 2024).
Knopman, D. S., H. Amieva, R. C. Petersen, G. Chetelat, D. M. Holtzman, B. T. Hyman, R. A. Nixon, and D. T. Jones. 2021. Alzheimer disease. Nature Reviews Disease Primers 7(1):33.
Kremer, I. 2024. Panelist Remarks. Committee on Research Priorities for Preventing and Treating Alzheimer’s Disease and Related Dementias Public Workshop, Hybrid. January 16–17, 2024.
Krishnan, K. J., T. E. Ratnaike, H. L. De Gruyter, E. Jaros, and D. M. Turnbull. 2012. Mitochondrial DNA deletions cause the biochemical defect observed in Alzheimer’s disease. Neurobiology and Aging 33(9):2210-2214.
Lamar, M., K. N. Kershaw, S. E. Leurgans, R. R. Mukherjee, B. S. Lange-Maia, D. X. Marquez, and L. L. Barnes. 2023. Neighborhood-level social vulnerability and individual-level cognitive and motor functioning over time in older non-Latino Black and Latino adults. Frontiers of Human Neuroscience 17:1125906.
Lamb, B. 2024. Panelist Remarks. Committee on Research Priorities for Preventing and Treating Alzheimer’s Disease and Related Dementias Public Workshop, Hybrid. January 16–17, 2024.
Lambert, J. C., A. Ramirez, B. Grenier-Boley, and C. Bellenguez. 2023. Step by step: Towards a better understanding of the genetic architecture of Alzheimer’s disease. Molecular Psychiatry 28:2716-2727.
Landau, S. M., D. Harvey, C. M. Madison, E. M. Reiman, N. L. Foster, P. S. Aisen, R. C. Petersen, L. M. Shaw, J. Q. Trojanowski, C. R. Jack, Jr., M. W. Weiner, and W. J. Jagust. 2010. Comparing predictors of conversion and decline in mild cognitive impairment. Neurology 20(75):230-238.
Lawlor, D. A., K. Tilling, and G. Davey Smith. 2016. Triangulation in aetiological epidemiology. International Journal of Epidemiology 45(6):1866-1886.
Lee, J. H., D. S. Yang, C. N. Goulbourne, E. Im, P. Stavrides, A. Pensalfini, H. Chan, C. Bouchet-Marquis, C. Bleiwas, M. J. Berg, C. Huo, J. Peddy, M. Pawlik, E. Levy, M. Rao, M. Staufenbiel, and R. A. Nixon. 2022. Faulty autolysosome acidification in Alzheimer’s disease mouse models induces autophagic build-up of Aβ in neurons, yielding senile plaques. Nature Neuroscience 25(6):688-701.
Lefevre-Arbogast, S., J. Chaker, F. Mercier, R. Barouki, X. Coumoul, G. W. Miller, A. David, and C. Samieri. 2024. Assessing the contribution of the chemical exposome to neurodegenerative disease. Nature Neuroscience 27(5):812-821.
Lehmann, D., A. Elshorbagy, and M. Hurley. 2023. Many paths to Alzheimer’s disease: A unifying hypothesis integrating biological, chemical and physical risk factors. Journal of Alzheimer’s Disease 95(4):1371-1382.
Liddelow, S. A., K. A. Guttenplan, L. E. Clarke, F. C. Bennett, C. J. Bohlen, L. Schirmer, M. L. Bennett, A. E. Munch, W. S. Chung, T. C. Peterson, D. K. Wilton, A. Frouin, B. A. Napier, N. Panicker, M. Kumar, M. S. Buckwalter, D. H. Rowitch, V. L. Dawson, T. M. Dawson, B. Stevens, and B. A. Barres. 2017. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541(7638):481-487.
Lim, Y., V. M. Kehm, E. B. Lee, J. H. Soper, C. Li, J. Q. Trojanowski, and V. M. Lee. 2011. Alpha-syn suppression reverses synaptic and memory defects in a mouse model of dementia with Lewy bodies. Journal of Neuroscience 31(27):10076-10087.
Lima, T., T. Y. Li, A. Mottis, and J. Auwerx. 2022. Pleiotropic effects of mitochondria in aging. Nature Aging 2(3):199-213.
Lin, Y. F., L. Y. Wang, C. S. Chen, C. C. Li, and Y. H. Hsiao. 2021. Cellular senescence as a driver of cognitive decline triggered by chronic unpredictable stress. Neurobiology Stress 15:100341.
Lin, P. B., A. P. Tsai, D. Soni, A. Lee-Gosselin, M. Moutinho, S. S. Puntambekar, G. E. Landreth, B. T. Lamb, and A. L. Oblak. 2023. INPP5D deficiency attenuates amyloid pathology in a mouse model of Alzheimer’s disease. Alzheimer’s & Dementia 19(6):2528-2537.
Lindbohm, J. V., N. Mars, P. N. Sipilä, A. Singh-Manoux, H. Runz; FinnGen; G. Livingston, S. Seshadri, R. Xavier, A. D. Hingorani, S. Ripatti, and M. Kivimäki. 2022. Immune system-wide Mendelian randomization and triangulation analyses support autoimmunity as a modifiable component in dementia-causing diseases. Nature Aging 2(10):956-972.
Liu, F., J. Xu, L. Guo, W. Qin, M. Liang, G. Schumann, and C. Yu. 2023a. Environmental neuroscience linking exposome to brain structure and function underlying cognition and behavior. Molecular Psychiatry 28(1):17-27.
Liu, N., X. Jiang, and H. Li. 2023b. The viral hypothesis in Alzheimer’s disease: SARS-CoV-2 on the cusp. Frontiers in Aging and Neuroscience 15:1129640.
Livingston, G., J. Huntley, K. Y. Liu, S. G. Costafreda, G. Selbæk, S. Alladi, D. Ames, S. Banerjee, A. Burns, C. Brayne, N. C. Fox, C. P. Ferri, L. N. Gitlin, R. Howard, H. C. Kales, M. Kivimäki, E. B. Larson, N. Nakasujja, K. Rockwood, Q. Samus, K. Shirai, A. Singh-Manoux, L. S. Schneider, S. Walsh, Y. Yao, A. Sommerlad, and N. Mukadam. 2024. Dementia prevention, intervention, and care: 2024 Report of the Lancet Standing Commission. Lancet 404(10452):572-628.
Loeffler, D. A. 2019. Influence of normal aging on brain autophagy: A complex scenario. Frontiers in Aging and Neuroscience 11:49.
López-Otín, C., M. A. Blasco, L. Partridge, M. Serrano, and G. Kroemer. 2023. Hallmarks of aging: An expanding universe. Cell 186(2):243-278.
Lourida, I., E. Hannon, T. J. Littlejohns, K. M. Langa, E. Hypponen, E. Kuzma, and D. J. Llewellyn. 2019. Association of lifestyle and genetic risk with incidence of dementia. JAMA 322(5):430-437.
Lutshumba, J., B. Nikolajczyk, and A. Bachstetter. 2021. Dysregulation of systemic immunity in aging and dementia. Frontiers in Cellular Neuroscience 15:652111.
Magen, I., and M. F. Chesselet. 2011. Mouse models of cognitive deficits due to alpha-synuclein pathology. Journal of Parkinson’s Disease 1(3):217-227.
Maloney, B., and D. Lahiri. 2016. Epigenetics of dementia: Understanding the disease as a transformation rather than a state. Lancet 15(7):760-774.
Manzoni, C., D. A. Kia, R. Ferrari, G. Leonenko, B. Costa, V. Saba, E. Jabbari, M. M. Tan, D. Albani, V. Alvarez, I. Alvarez, O. A. Andreassen, A. Angiolillo, A. Arighi, M. Baker, L. Benussi, V. Bessi, G. Binetti, D. J. Blackburn, M. Boada, B. F. Boeve, S. Borrego-Ecija, B. Borroni, G. Brathen, W. S. Brooks, A. C. Bruni, P. Caroppo, S. Bandres-Ciga, J. Clarimon, R. Colao, C. Cruchaga, A. Danek, S. C. de Boer, I. de Rojas, A. di Costanzo, D. W. Dickson, J. Diehl-Schmid, C. Dobson-Stone, O. Dols-Icardo, A. Donizetti, E. Dopper, E. Durante, C. Ferrari, G. Forloni, F. Frangipane, L. Fratiglioni, M. G. Kramberger, D. Galimberti, M. Gallucci, P. Garcia-Gonzalez, R. Ghidoni, G. Giaccone, C. Graff, N. R. Graff-Radford, J. Grafman, G. M. Halliday, D. G. Hernandez, L. E. Hjermind, J. R. Hodges, G. Holloway, E. D. Huey, I. Illan-Gala, K. A. Josephs, D. S. Knopman, M. Kristiansen, J. B. Kwok, I. Leber, H. L. Leonard, I. Libri, A. Lleo, I. R. Mackenzie, G. K. Madhan, R. Maletta, M. Marquie, A. Maver, M. Menendez-Gonzalez, G. Milan, B. L. Miller, C. M. Morris, H. R. Morris, B. Nacmias, J. Newton, J. E. Nielsen, C. Nilsson, V. Novelli, A. Padovani, S. Pal, F. Pasquier, P. Pastor, R. Perneczky, B. Peterlin, R. C. Petersen, O. Piguet, Y. A. Pijnenburg, A. A. Puca, R. Rademakers, I. Rainero, L. M. Reus, A. M. Richardson, M. Riemenschneider, E. Rogaeva, B. Rogelj, S. Rollinson, H. Rosen, G. Rossi, J. B. Rowe, E. Rubino, A. Ruiz, E. Salvi, R. Sanchez-Valle, S. B. Sando, A. F. Santillo, J. A. Saxon, J. C. Schlachetzki, S. W. Scholz, H. Seelaar, W. W. Seeley, M. Serpente, S. Sorbi, S. Sordon, P. St George-Hyslop, J. C. Thompson, C. Van Broeckhoven, V. M. Van Deerlin, S. J. Van der Lee, J. Van Swieten, F. Tagliavini, J. van der Zee, A. Veronesi, E. Vitale, M. L.
Waldo, J. S. Yokoyama, M. A. Nalls, P. Momeni, A. B. Singleton, J. Hardy, and V. Escott-Price. 2024. Genome-wide analyses reveal a potential role for the MAPT, MOBP, and APOE loci in sporadic frontotemporal dementia. American Journal of Human Genetics 111(7):1316-1329.
Marlies Oosthoek, L. V., A. de Wilde, B. Bongers, D. Antwi-Berko, P. Scheltens, P. van Bokhoven, E. G. B. Vijverberg, and C. E. Teunissen. 2024. Utilization of fluid-based biomarkers as endpoints in disease-modifying clinical trials for Alzheimer’s disease: A systematic review. Alzheimer’s Research & Therapy 16:93.
Marquez, F., W. Tarraf, A. M. Stickel, K. A. Gonzalez, F. D. Testai, J. Cai, L. C. Gallo, G. A. Talavera, M. L. Daviglus, S. Wassertheil-Smoller, C. DeCarli, N. Schneiderman, and H. M. Gonzalez. 2024. Hypertension, cognitive decline, and mild cognitive impairment among diverse Hispanics/Latinos: Study of Latinos—Investigation of Neurocognitive Aging Results (SOL-INCA). Journal of Alzheimer’s Disease 97(3):1449-1461.
Marsit, C. J. 2015. Influence of environmental exposure on human epigenetic regulation. Journal of Experimental Biology 218(Pt 1):71-79.
Martinez-Feduchi, P., P. Jin, and B. Yao. 2024. Epigenetic modifications of DNA and RNA in Alzheimer’s disease. Frontiers in Molecular Neuroscience 17:1398026.
Matthay, E. C., E. Hagan, L. M. Gottlieb, M. L. Tan, D. Vlahov, N. E. Adler, and M. M. Glymour. 2020. Alternative causal inference methods in population health research: Evaluating tradeoffs and triangulating evidence. SSM Population Health 10:100526.
Mayne, K., J. A. White, C. E. McMurran, F. J. Rivera, and A. G. de la Fuente. 2020. Aging and neurodegenerative disease: Is the adaptive immune system a friend or foe? Frontiers in Aging Neuroscience 12:572090.
Mehta, R. I., and R. I. Mehta. 2023. The vascular-immune hypothesis of Alzheimer’s disease. Biomedicines 11(2):408.
Mei, Z., G. Liu, B. Zhao, Z. He, and S. Gu. 2023. Emerging roles of epigenetics in lead-induced neurotoxicity. Environment International 181:108253.
Melcher, E. M., L. Vilen, A. Pfaff, S. Lim, A. DeWitt, W. R. Powell, B. B. Bendlin, and A. J. H. Kind. 2024. Deriving life-course residential histories in brain bank cohorts: A feasibility study. Alzheimer’s & Dementia 20(5):3219-3227.
Mensikova, K., R. Matej, C. Colosimo, R. Rosales, L. Tuckova, J. Ehrmann, D. Hrabos, K. Kolarikova, R. Vodicka, R. Vrtel, M. Prochazka, M. Nevrly, M. Kaiserova, S. Kurcova, P. Otruba, and P. Kanovsky. 2022. Lewy body disease or diseases with Lewy bodies? NPJ Parkinsons Disease 8(1):3.
Mez, J., J. Chung, G. Jun, J. Kriegel, A. P. Bourlas, R. Sherva, M. W. Logue, L. L. Barnes, D. A. Bennett, J. D. Buxbaum, G. S. Byrd, P. K. Crane, N. Ertekin-Taner, D. Evans, M. D. Fallin, T. Foroud, A. Goate, N. R. Graff-Radford, K. S. Hall, M. I. Kamboh, W. A. Kukull, E. B. Larson, J. J. Manly, J. L. Haines, R. Mayeux, M. A. Pericak-Vance, G. D. Schellenberg, K. L. Lunetta, and L. A. Farrer. 2017. Two novel loci, COBL and SLC10A2, for Alzheimer’s disease in African Americans. Alzheimer’s & Dementia 13(2):119-129.
Mielke, M. 2024. Sex and gender differences in Alzheimer’s disease and Alzheimer’s disease related dementias. Commissioned Paper for the Committee on Research Priorities for Preventing and Treating Alzheimer’s Disease and Related Dementias.
Migliore, L., and F. Coppedè. 2022. Gene–environment interactions in Alzheimer disease: The emerging role of epigenetics. Nature Reviews Neurology 18:643-660.
Miller, G. 2024. Presentation at Committee on Research Priorities for Preventing and Treating Alzheimer’s Disease and Related Dementias Public Workshop, Hybrid. January 16–17, 2024.
Miller, M. B., A. Y. Huang, J. Kim, Z. Zhou, S. L. Kirkham, E. A. Maury, J. S. Ziegenfuss, H. C. Reed, J. E. Neil, L. Rento, S. C. Ryu, C. C. Ma, L. J. Luquette, H. M. Ames, D. H. Oakley, M. P. Frosch, B. T. Hyman, M. A. Lodato, E. A. Lee, and C. A. Walsh. 2022. Somatic genomic changes in single Alzheimer’s disease neurons. Nature 604(7907):714-722.
Miyashita, A., A. Koike, G. Jun, L. S. Wang, S. Takahashi, E. Matsubara, T. Kawarabayashi, M. Shoji, N. Tomita, H. Arai, T. Asada, Y. Harigaya, M. Ikeda, M. Amari, H. Hanyu, S. Higuchi, T. Ikeuchi, M. Nishizawa, M. Suga, Y. Kawase, H. Akatsu, K. Kosaka, T. Yamamoto, M. Imagawa, T. Hamaguchi, M. Yamada, T. Morihara, M. Takeda, T. Takao, K. Nakata, Y. Fujisawa, K. Sasaki, K. Watanabe, K. Nakashima, K. Urakami, T. Ooya, M. Takahashi, T. Yuzuriha, K. Serikawa, S. Yoshimoto, R. Nakagawa, J. W. Kim, C. S. Ki, H. H. Won, D. L. Na, S. W. Seo, I. Mook-Jung, P. St George-Hyslop, R. Mayeux, J. L. Haines, M. A. Pericak-Vance, M. Yoshida, N. Nishida, K. Tokunaga, K. Yamamoto, S. Tsuji, I. Kanazawa, Y. Ihara, G. D. Schellenberg, L. A. Farrer, and R. Kuwano. 2013. SORL1 is genetically associated with late-onset Alzheimer’s disease in Japanese, Koreans and Caucasians. PLoS ONE 8(4):e58618.
Miyashita, A., M. Kikuchi, N. Hara, and T. Ikeuchi. 2023. Genetics of Alzheimer’s disease: An East Asian perspective. Journal of Human Genetics 68(3):115-124.
MODEL-AD. 2024. MODEL-AD. https://www.model-ad.org/ (accessed August 22, 2024).
Morawe, T., C. Hiebel, A. Kern, and C. Behl. 2012. Protein homeostasis, aging and Alzheimer’s disease. Molecular Neurobiology 46(1):41-54.
Musi, N., J. M. Valentine, K. R. Sickora, E. Baeuerle, C. S. Thompson, Q. Shen, and M. E. Orr. 2018. Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell 17(6):e12840.
NASEM (National Academies of Sciences, Engineering, and Medicine). 2018. Future directions for the demography of aging: Proceedings of a workshop. Edited by M. K. Majmundar and M. D. Hayward. Washington, DC: The National Academies Press.
NASEM. 2020a. Environmental neuroscience: Advancing the understanding of how chemical exposures impact brain health and disease: Proceedings of a workshop. Edited by C. Stroud, S. M. Posey Norris and L. Bain. Washington, DC: The National Academies Press.
NASEM. 2020b. Integrating the science of aging and environmental health research: Proceedings of a workshop—in brief. Edited by K. Sawyer, F. Sharples and R. Poole. Washington, DC: The National Academies Press.
NASEM. 2023. Nonhuman primate models in biomedical research: State of the science and future needs. Edited by O. C. Yost, A. Downey and K. S. Ramos. Washington, DC: The National Academies Press.
NASEM. 2024. Preventing and treating Alzheimer’s disease and related dementias: Promising research and opportunities to accelerate progress: Proceedings of a workshop—in brief. Edited by A. Downey and O. Yost. Washington, DC: The National Academies Press.
Nativio, R., G. Donahue, A. Berson, Y. Lan, A. Amlie-Wolf, F. Tuzer, J. B. Toledo, S. J. Gosai, B. D. Gregory, C. Torres, J. Q. Trojanowski, L. S. Wang, F. B. Johnson, N. M. Bonini, and S. L. Berger. 2018. Dysregulation of the epigenetic landscape of normal aging in Alzheimer’s disease. Nature Neuroscience 21(4):497-505.
Nativio, R., Y. Lan, G. Donahue, S. Sidoli, A. Berson, A. R. Srinivasan, O. Shcherbakova, A. Amlie-Wolf, J. Nie, X. Cui, C. He, L. S. Wang, B. A. Garcia, J. Q. Trojanowski, N. M. Bonini, and S. L. Berger. 2020. An integrated multi-omics approach identifies epigenetic alterations associated with Alzheimer’s disease. Nature Genetics 52(10):1024-1035.
Naudi, A., R. Cabre, M. Jove, V. Ayala, H. Gonzalo, M. Portero-Otin, I. Ferrer, and R. Pamplona. 2015. Lipidomics of human brain aging and Alzheimer’s disease pathology. International Review of Neurobiology 122:133-189.
Neuner, S. M., M. Telpoukhovskaia, V. Menon, K. M. S. O’Connell, T. J. Hohman, and C. C. Kaczorowski. 2022. Translational approaches to understanding resilience to Alzheimer’s disease. Trends in neurosciences 45(5):369–383.
NIA (National Insitute on Aging). 2016. Decoding the molecular ties between vascular disease and Alzheimer’s. https://www.nia.nih.gov/news/decoding-molecular-ties-between-vascular-disease-and-alzheimers (accessed October 17, 2024).
NIA. 2019. NIH-funded translational research centers to speed, diversify Alzheimer’s drug discovery. https://www.nia.nih.gov/news/nih-funded-translational-research-centers-speed-diversify-alzheimers-drug-discovery#:~:text=To%20help%20meet%20the%20urgent,over%20the%20next%20five%20years (accessed October 18, 2024).
NIA. 2020. Understanding the role of the exposome in brain aging, Alzheimer’s disease (AD) and AD-related dementias (ADRD). Baltimore, MD: NIA.
NIA. 2022a. Microphysiological systems to advance precision medicine for Alzheimer’s disease (AD) and AD-related dementias (ADRD) treatment and prevention. Baltimore, MD: NIA.
NIA. 2022b. Accelerating Medicines Partnership program for Alzheimer’s Disease (AMP AD 2.0). https://www.nia.nih.gov/research/amp-ad-second-iteration (accessed August 19, 2024).
NIA. 2023a. Exploring the role of the exposome in aging and age-related diseases. https://www.nia.nih.gov/exposome (accessed August 16, 2024).
NIA. 2023b. Research enterprise. https://www.nia.nih.gov/report-2019-2020-scientific-advances-prevention-treatment-and-care-dementia/research-enterprise (accessed August 22, 2024).
NIA. 2023c. TREAT-AD centers: Open source tools to reinvigorate Alzheimer’s drug discovery. https://www.nia.nih.gov/research/blog/2023/12/treat-ad-centers-open-source-tools-reinvigorate-alzheimers-drug-discovery (accessed August 19, 2024).
NIA. 2023d. Fiscal year 2024 budget. https://www.nia.nih.gov/about/budget/fiscal-year-2024-budget (accessed August 22, 2024).
NIA. 2024a. Alzheimer Biomarkers Consortium—Down Syndrome (ABC-DS). https://www.nia.nih.gov/research/abc-ds (accessed August 23, 2024).
NIA. 2024b. Alzheimer’s disease sequencing project consortia. https://www.nia.nih.gov/research/dn/alzheimers-disease-sequencing-project-consortia (accessed August 22, 2024).
NIA. 2024c. Successful Trajectories of Aging: Reserve and Resilience in Rats (STARRRS). https://www.nia.nih.gov/research/labs/about-irp/successful-trajectories-of-aging-reserve-and-resilience-in-rats#:~:text=About%20STARRRS,-Aging%20in%20humans&text=In%20contrast%2C%20other%20people%20experience,of%20neurocognitive%20aging%20in%20rats (accessed October 23, 2024).
NIA. 2024d. Accelerating Medicines Partnership Program for Alzheimer’s Disease (AMP AD). https://www.nia.nih.gov/research/amp-ad (accessed August 22, 2024).
NIA. 2024e. 2024 NIH Alzheimer’s Research Summit: Building a precision medicine research enterprise. https://www.nia.nih.gov/2024-alzheimers-summit (accessed October 25, 2024).
NIA. 2024f. Fiscal year 2025 budget. https://www.nia.nih.gov/about/budget/fiscal-year-2025-budget (accessed August 22, 2024).
NIH (National Insitutes of Health). 2015. Consideration of sex as a biological variable in NIH-funded research. https://grants.nih.gov/grants/guide/notice-files/not-od-15-102.html (accessed October 23, 2024).
NIH. 2019. New NIH-funded translational research centers to speed, diversify Alzheimer’s drug discovery. https://www.nih.gov/news-events/news-releases/new-nih-funded-translational-research-centers-speed-diversify-alzheimers-drug-discovery (accessed August 22, 2024).
NIH. 2023. Center for exposome research coordination to accelerate precision environmental health. https://grants.nih.gov/grants/guide/rfa-files/RFA-ES-23-010.html (accessed August 16, 2024).
NIH. 2024. Celluar Senescence Network (SenNet). https://commonfund.nih.gov/senescence (accessed August 19, 2024).
Nikolac Perkovic, M., A. Videtic Paska, M. Konjevod, K. Kouter, D. Svob Strac, G. Nedic Erjavec, and N. Pivac. 2021. Epigenetics of Alzheimer’s disease. Biomolecules 11(2):195.
NINDS (National Institute of Neurological Disorders and Stroke). 2024a. The neural exposome. https://www.ninds.nih.gov/current-research/research-funded-ninds/translational-research/onetox-neural-exposome-and-toxicology-programs/neural-exposome (accessed August 16, 2024).
NINDS. 2024b. Accelerating Medicines Partnership® for Amyotrophic Lateral Sclerosis (AMP® ALS). https://www.ninds.nih.gov/current-research/focus-disorders/amyotrophic-lateral-sclerosis/accelerating-medicines-partnershipr-amyotrophic-lateral-sclerosis-ampr-als (accessed October 24, 2024).
Nixon, R. A. 2013. The role of autophagy in neurodegenerative disease. Nature Medicine 19(8):983-997.
Nixon, R. A. 2020. The aging lysosome: An essential catalyst for late-onset neurodegenerative diseases. Biochimica et Biophysica Acta Proteins & Proteomics 1868(9):140443.
Noche, J. A., H. Radhakrishnan, M. F. Ubele, K. Boaz, J. L. Mefford, E. D. Jones, H. Y. van Rooyen, J. A. Perpich, K. McCarty, B. Meacham, J. Smiley, S. A. Bembenek Bailey, L. G. Puskas, D. K. Powell, L. Sordo, M. J. Phelan, C. M. Norris, E. Head, and C. E. L. Stark. 2024. Age-related brain atrophy and the positive effects of behavioral enrichment in middle-aged beagles. Journal of Neuroscience 44(20):e2366232024.
Oblak, A. L., S. Forner, P. R. Territo, M. Sasner, G. W. Carter, G. R. Howell, S. J. Sukoff-Rizzo, B. A. Logsdon, L. M. Mangravite, A. Mortazavi, D. Baglietto-Vargas, K. N. Green, G. R. MacGregor, M. A. Wood, A. J. Tenner, F. M. LaFerla, B. T. Lamb, and the MODEL-AD and Consortium. 2020. Model organism development and evaluation for late-onset Alzheimer’s disease: MODEL-AD. Alzheimer’s & Dementia (N Y) 6(1):e12110.
Ogrodnik, M., Y. Zhu, L. G. P. Langhi, T. Tchkonia, P. Kruger, E. Fielder, S. Victorelli, R. A. Ruswhandi, N. Giorgadze, T. Pirtskhalava, O. Podgorni, G. Enikolopov, K. O. Johnson, M. Xu, C. Inman, A. K. Palmer, M. Schafer, M. Weigl, Y. Ikeno, T. C. Burns, J. F. Passos, T. von Zglinicki, J. L. Kirkland, and D. Jurk. 2019. Obesity-induced cellular senescence drives anxiety and impairs neurogenesis. Cell Metabolism 29(5):1061-1077.e8.
Oppenheim, H. A., J. Lucero, and A. Guyot. 2013. Exposure to vehicle emissions results in altered blood brain barrier permeability and expression of matrix metalloproteinases and tight junction proteins in mice. Particle and Fibre Toxicology 10:62.
Orme, T., R. Guerreiro, and J. Bras. 2018. The genetics of dementia with Lewy bodies: Current understanding and future directions. Current Neurology and Neuroscience Reports 18:67.
Owaki, A., A. Tanaka, K. Furuhashi, Y. Watanabe, E. Koshi-Ito, T. Imaizumi, and S. Maruyama. 2024. Prognosis of microscopic polyangiitis is well predictable in the first 2 weeks of treatment. Clinical Experiments in Nephrology 28(7):701-706.
Pacholko, A., and C. Iadecola. 2024. Hypertension, neurodegeneration, and cognitive decline. Hypertension 81(5):991-1007.
Padmanabhan, P., and J. Gotz. 2023. Clinical relevance of animal models in aging-related dementia research. Nature Aging 3(5):481-493.
PAHO/WHO (Pan American Health Organization/World Health Organization). 2024. Healthy aging. https://www.paho.org/en/healthy-aging (accessed August 16, 2024).
Parrish, M. C., Y. J. Tan, K. V. Grimes, and D. Mochly-Rosen. 2019. Surviving in the valley of death: Opportunities and challenges in translating academic drug discoveries. Annual Review of Pharmacology and Toxicology 59:405-421.
Paşca, S. 2019. Assembling human brain organoids. Science 363(6423):126-127.
Patrick, K. L., S. L. Bell, C. G. Weindel, and R. O. Watson. 2019. Exploring the “multiple-hit hypothesis” of neurodegenerative disease: Bacterial infection comes up to bat. Frontiers in Cellular and Infection Microbiology 9:138.
Patten, K. T., and P. J. Lein. 2019. Gene-environment interactions determine risk for dementia: The influence of lifestyle on genetic risk for dementia. Annals of Translational Medicine 7(Suppl 8):S322.
Paulson, H. L., and I. Igo. 2011. Genetics of dementia. Seminal Neurology 31(5):449-460.
Pearl, J. 2009. Causality: Models, Reasoning, and Inference. Cambridge, UK: Cambridge University Press.
Peleg, S., C. Feller, A. G. Ladurner, and A. Imhof. 2016. The metabolic impact on histone acetylation and transcription in ageing. Trends in Biochemical Sciences 41(8):700-711.
Pellegrini, C., C. Pirazzini, C. Sala, L. Sambati, I. Yusipov, A. Kalyakulina, F. Ravaioli, K. M. Kwiatkowska, D. F. Durso, M. Ivanchenko, D. Monti, R. Lodi, C. Franceschi, P. Cortelli, P. Garagnani, and M. G. Bacalini. 2021. A meta-analysis of brain DNA methylation across sex, age, and Alzheimer’s disease points for accelerated epigenetic aging in neurodegeneration. Frontiers in Aging Neuroscience 13:639428.
Pericak-Vance, M. 2024. Alzheimer’s Disease Sequencing Project (ADSP). Presentation at Committee on Research Priorities for Preventing and Treating Alzheimer’s Disease and Related Dementias Public Workshop, Hybrid. January 16–17, 2024.
Peters, R., N. Ee, J. Peters, A. Booth, I. Mudway, and K. J. Anstey. 2019. Air pollution and dementia: A systematic review. Journal of Alzheimer’s Disease 70(S1):S145-S163.
Pham, T. N. M., N. Perumal, C. Manicam, M. Basoglu, S. Eimer, D. C. Fuhrmann, C. U. Pietrzik, A. M. Clement, H. Korschgen, J. Schepers, and C. Behl. 2023. Adaptive responses of neuronal cells to chronic endoplasmic reticulum (ER) stress. Redox Biology 67:102943.
Pilling, L. C., R. Joehanes, D. Melzer, L. W. Harries, W. Henley, J. Dupuis, H. Lin, M. Mitchell, D. Hernandez, S. X. Ying, K. L. Lunetta, E. J. Benjamin, A. Singleton, D. Levy, P. Munson, J. M. Murabito, and L. Ferrucci. 2015. Gene expression markers of age-related inflammation in two human cohorts. Experimental Gerontology 70:37-45.
Powell, W. R., W. R. Buckingham, J. L. Larson, L. Vilen, M. Yu, M. S. Salamat, B. B. Bendlin, R. A. Rissman, and A. J. H. Kind. 2020. Association of neighborhood-level disadvantage with Alzheimer disease neuropathology. JAMA Network Open 3(6):e207559.
Quach, A., M. E. Levine, T. Tanaka, A. T. Lu, B. H. Chen, L. Ferrucci, B. Ritz, S. Bandinelli, M. L. Neuhouser, J. M. Beasley, L. Snetselaar, R. B. Wallace, P. S. Tsao, D. Absher, T. L. Assimes, J. D. Stewart, Y. Li, L. Hou, A. A. Baccarelli, E. A. Whitsel, and S. Horvath. 2017. Epigenetic clock analysis of diet, exercise, education, and lifestyle factors. Aging 9(2):419-446.
Rabinovici, G. D., M. C. Carrillo, M. Forman, S. DeSanti, D. S. Miller, N. Kozauer, R. C. Petersen, C. Randolph, D. S. Knopman, E. E. Smith, M. Isaac, N. Mattsson, L. J. Bain, J. A. Hendrix, and J. R. Sims. 2017. Multiple comorbid neuropathologies in the setting of Alzheimer’s disease neuropathology and implications for drug development. Alzheimer’s & Dementia (N Y) 3(1):83-91.
Ramesh, M., P. Gopinath, and T. Govindaraju. 2019. Role of post-translational modifications in Alzheimer’s disease. ChemBioChem 21(8):1052-1079.
Raulin, A.-C., S. V. Doss, Z. A. Trottier, T. C. Ikezu, G Bu, and C.-C. Liu. 2022. ApoE in Alzheimer’s disease: Pathophysiology and therapeutic strategies. Molecular Neurodegeneration 17(1):72.
Reitz, C., G. Jun, A. Naj, R. Rajbhandary, B. N. Vardarajan, L.-S. Wang, O. Valladares, C.-F. Lin, E. B. Larson, N. R. Graff-Radford, D. Evans, P. L. De Jager, P. K. Crane, J. D. Buxbaum, J. R. Murrell, T. Raj, N. Ertekin-Taner, M. Logue, C. T. Baldwin, R. C. Green, L. L. Barnes, L. B. Cantwell, M. D. Fallin, R. C. P. Go, P. Griffith, T. O. Obisesan, J. J. Manly, K. L. Lunetta, M. I. Kamboh, O. L. Lopez, D. A. Bennett, H. Hendrie, K. S. Hall, A. M. Goate, G. S. Byrd, W. A. Kukull, T. M. Foroud, J. L. Haines, L. A. Farrer, M. A. Pericak-Vance, G. D. Schellenberg, R. Mayeux, and Alzheimer Disease Genetics Consortium. 2013. Variants in the ATP-binding cassette transporter (ABCA7), Apolipoprotein E ϵ4, and the risk of late-onset Alzheimer disease in African Americans. JAMA 309(14):1483-1492.
Reitz, C., M. A. Pericak-Vance, T. Foroud, and R. Mayeux. 2023. A global view of the genetic basis of Alzheimer’s disease. Nature Reviews Neurology 19(5):261-277.
Reuben, A., L. S. Richmond-Rakerd, B. Milne, D. Shah, A. Pearson, S. Hogan, D. Ireland, R. Keenan, A. R. Knodt, T. Melzer, R. Poulton, S. Ramrakha, E. T. Whitman, A. R. Hariri, T. E. Moffitt, and A. Caspi. 2024. Dementia, dementia’s risk factors and premorbid brain structure are concentrated in disadvantaged areas: National register and birth-cohort geographic analyses. Alzheimer’s & Dementia 20(5):3167-3178.
Roberson, E. D. 2012. Mouse models of frontotemporal dementia. Annals of Neurology 72(6):837-849.
Robinson, J. L., H. Richardson, S. X. Xie, E. Suh, V. M. Van Deerlin, B. Alfaro, N. Loh, M. Porras-Paniagua, J. J. Nirschl, D. Wolk, V. M. Lee, E. B. Lee, and J. Q. Trojanowski. 2021. The development and convergence of co-pathologies in Alzheimer’s disease. Brain 144(3):953-962.
Rongve, A., A. Witoelar, A. Ruiz, L. Athanasiu, C. Abdelnour, J. Clarimon, S. Heilmann-Heimbach, I. Hernández, S. Moreno-Grau, I. de Rojas, E. Morenas-Rodríguez, T. Fladby, S. B. Sando, G. Bråthen, F. Blanc, O. Bousiges, A. W. Lemstra, I. van Steenoven, E. Londos, I. S. Almdahl, L. Pålhaugen, J. A. Eriksen, S. Djurovic, E. Stordal, I. Saltvedt, I. D. Ulstein, F. Bettella, R. S. Desikan, A. V. Idland, M. Toft, L. Pihlstrøm, J. Snaedal, L. Tárraga, M. Boada, A. Lleó, H. Stefánsson, K. Stefánsson, A. Ramírez, D. Aarsland, and O. A. Andreassen. 2019. GBA and APOE ε4 associate with sporadic dementia with Lewy bodies in European genome wide association study. Scientific Reports 9(1):7013.
Rozek, L. S., D. C. Dolinoy, M. A. Sartor, and G. S. Omenn. 2014. Epigenetics: Relevance and implications for public health. Annual Review of Public Health 35:105-122.
Rubinsztein, D. C., G. Marino, and G. Kroemer. 2011. Autophagy and aging. Cell 146(5):682-695.
Ryu, J. K., and J. G. McLarnon. 2009. A leaky blood-brain barrier, fibrinogen infiltration and microglial reactivity in inflamed Alzheimer’s disease brain. Journal of Cellular and Molecular Medicine 13(9A):2911-2925.
Safarlou, C. W., K. R. Jongsma, R. Vermeulen, and A. L. Bredenoord. 2023. The ethical aspects of exposome research: A systematic review. Exposome 3(1):osad004.
Salter, M. W., and B. Stevens. 2017. Microglia emerge as central players in brain disease. Nature Medicine 23(9):1018-1027.
Sampson, T. R., J. W. Debelius, T. Thron, S. Janssen, G. G. Shastri, Z. E. Ilhan, C. Challis, C. E. Schretter, S. Rocha, V. Gradinaru, M. F. Chesselet, A. Keshavarzian, K. M. Shannon, R. Krajmalnik-Brown, P. Wittung-Stafshede, R. Knight, and S. K. Mazmanian. 2016. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell 167(6):1469-1480 e1412.
Sander, M., J. Brown, K. Lunnon, and C. Ballard. 2023. Characterising the D409V/WT mouse as a novel model of Lewy body dementia. Alzheimer’s & Dementia 19(S12).
Scheltens, P., B. De Strooper, M. Kivipelto, H. Holstege, G. Chételat, C. E. Teunissen, J. Cummings, and W. M. van der Flier. 2021. Alzheimer’s disease. Lancet 397(10284):1577-1590.
Schrott, R., A. Song, and C. Ladd-Acosta. 2022. Epigenetics as a biomarker for early-life environmental exposure. Current Environmental Health Reports 9:604-624.
Schumacher, B., J. Pothof, J. Vijg, and J. H. J. Hoeijmakers. 2021. The central role of DNA damage in the ageing process. Nature 592(7856):695-703.
Scrivo, A., M. Bourdenx, O. Pampliega, and A. M. Cuervo. 2018. Selective autophagy as a potential therapeutic target for neurodegenerative disorders. Lancet Neurology 17(9):802-815.
SenNet Consortium. 2022. NIH SenNet consortium to map senescent cells throughout the human lifespan to understand physiological health. Nature Aging 2(12):1090-1100.
Seok, J., H. S. Warren, A. G. Cuenca, M. N. Mindrinos, H. V. Baker, W. Xu, D. R. Richards, G. P. McDonald-Smith, H. Gao, L. Hennessy, C. C. Finnerty, C. M. Lopez, S. Honari, E. E. Moore, J. P. Minei, J. Cuschieri, P. E. Bankey, J. L. Johnson, J. Sperry, A. B. Nathens, T. R. Billiar, M. A. West, M. G. Jeschke, M. B. Klein, R. L. Gamelli, N. S. Gibran, B. H. Brownstein, C. Miller-Graziano, S. E. Calvano, P. H. Mason, J. P. Cobb, L. G. Rahme, S. F. Lowry, R. V. Maier, L. L. Moldawer, D. N. Herndon, R. W. Davis, W. Xiao, R. G. Tompkins, and the Inflammation and Host Response to Injury, Large Scale Collaborative Research Program. 2013. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proceedings of the National Academy of Sciences USA 110(9):3507-3512.
Shen, Q., K. T. Patten, A. Valenzuela, P. J. Lein, and A. Y. Taha. 2022. Probing changes in brain esterified oxylipin concentrations during the early stages of pathogenesis in Alzheimer’s disease transgenic rats. Neuroscience Letters 791:136921.
Shi, L., K. Steenland, H. Li, P. Liu, Y. Zhang, R. H. Lyles, W. J. Requia, S. D. Ilango, H. H. Chang, T. Wingo, R. J. Weber, and J. Schwartz. 2021. A national cohort study (2000-2018) of long-term air pollution exposure and incident dementia in older adults in the United States. Nature Communication 12(1):6754.
Shigemizu, D., R. Mitsumori, S. Akiyama, A. Miyashita, T. Morizono, S. Higaki, Y. Asanomi, N. Hara, G. Tamiya, K. Kinoshita, T. Ikeuchi, S. Niida, and K. Ozaki. 2021. Ethnic and trans-ethnic genome-wide association studies identify new loci influencing Japanese Alzheimer’s disease risk. Translational Psychiatry 11(1):151.
Shih, R. A., H. Hu, M. G. Weisskopf, and B. S. Schwartz. 2007. Cumulative lead dose and cognitive function in adults: A review of studies that measured both blood lead and bone lead. Environmental Health Perspectives 115(3):483-492.
Shireby, G. L., J. P. Davies, P. T. Francis, J. Burrage, E. M. Walker, G. W. A. Neilson, A. Dahir, A. J. Thomas, S. Love, R. G. Smith, K. Lunnon, M. Kumari, L. C. Schalkwyk, K. Morgan, K. Brookes, E. Hannon, and J. Mill. 2020. Recalibrating the epigenetic clock: Implications for assessing biological age in the human cortex. Brain 143(12):3763-3775.
Sierra, F. 2016. The emergence of geroscience as an interdisciplinary approach to the enhancement of health span and life span. Cold Spring Harbor Perspectives in Medicine 6(4):a025163.
Sierra, F., A. Caspi, R. H. Fortinsky, L. Haynes, G. J. Lithgow, T. E. Moffitt, S. J. Olshansky, D. Perry, E. Verdin, and G. A. Kuchel. 2021. Moving geroscience from the bench to clinical care and health policy. Journal of the American Geriatric Society 69(9):2455-2463.
Silberstein, M., N. Nesbit, J. Cai, and P. H. Lee. 2021. Pathway analysis for genome-wide genetic variation data: Analytic principles, latest developments, and new opportunities. Journal of Genetics and Genomics 48(3):173-183.
Sims, R., M. Hill, and J. Williams. 2020. The multiplex model of the genetics of Alzheimer’s disease. Nature Neuroscience 23(3):311-322.
Sinclair, D., A. J. Canty, J. M. Ziebell, A. Woodhouse, J. M. Collins, S. Perry, E. Roccati, M. Kuruvilla, J. Leung, R. Atkinson, J. C. Vickers, A. L. Cook, and A. E. King. 2024. Experimental laboratory models as tools for understanding modifiable dementia risk. Alzheimer’s & Dementia 20(6):4260-4289.
Sipilä, P. N., N. Heikkila, J. V. Lindbohm, C. Hakulinen, J. Vahtera, M. Elovainio, S. Suominen, A. Vaananen, A. Koskinen, S. T. Nyberg, J. Pentti, T. E. Strandberg, and M. Kivimaki. 2021. Hospital-treated infectious diseases and the risk of dementia: A large, multicohort, observational study with a replication cohort. Lancet Infectious Diseases 21(11):1557-1567.
Siroux, V., L. Agier, and R. Slama. 2016. The exposome concept: A challenge and a potential driver for environmental health research. European Respiratory Review 25(140):124-129.
Slanzi, A., G. Iannoto, B. Rossi, E. Zenaro, and G. Constantin. 2020. In vitro models of neurodegenerative diseases. Frontiers in Cell and Developmental Biology 8:328.
Smith, M. 2024. Panelist Remarks. Committee on Research Priorities for Preventing and Treating Alzheimer’s Disease and Related Dementias Public Workshop, Hybrid. January 16–17, 2024.
Steele, O. G., A. C. Stuart, L. Minkley, K. Shaw, O. Bonnar, S. Anderle, A. C. Penn, J. Rusted, L. Serpell, C. Hall, and S. King. 2022. A multi-hit hypothesis for an APOE4-dependent pathophysiological state. European Journal of Neuroscience 56(9):5476-5515.
Stephan, B. C. M., L. Cochrane, A. H. Kafadar, J. Brain, E. Burton, B. Myers, C. Brayne, A. Naheed, K. J. Anstey, A. W. Ashor, and M. Siervo. 2024. Population attributable fractions of modifiable risk factors for dementia: A systematic review and meta-analysis. Lancet Healthy Longevity 5(6):e406-e421.
Stern, Y., C. A. Barnes, C. Grady, R. N. Jones, and N. Raz. 2019. Brain reserve, cognitive reserve, compensation, and maintenance: Operationalization, validity, and mechanisms of cognitive resilience. Neurobiology and Aging 83:124-129.
Stern, Y., M. Albert, C. A. Barnes, R. Cabeza, A. Pascual-Leone, and P. R. Rapp. 2023. A framework for concepts of reserve and resilience in aging. Neurobiology and Aging 124:100-103.
Stites, S. D., S. Midgett, D. Mechanic-Hamilton, M. Zuelsdorff, C. M. Glover, D. X. Marquez, J. E. Balls-Berry, M. L. Streitz, G. Babulal, J. F. Trani, J. N. Henderson, L. L. Barnes, J. Karlawish, and D. A. Wolk. 2022. Establishing a framework for gathering structural and social determinants of health in Alzheimer’s disease research centers. Gerontologist 62(5):694-703.
Streit, W. J., and Q. S. Xue. 2014. Human CNS immune senescence and neurodegeneration. Current Opinion in Immunology 29:93-96.
Stylianou, M., B. Zaaimi, A. Thomas, J. P. Taylor, and F. E. N. LeBeau. 2020. Early disruption of cortical sleep-related oscillations in a mouse model of dementia with Lewy bodies (DLB) expressing human mutant (A30P) alpha-synuclein. Frontiers in Neuroscience 14:579867.
Sukoff Rizzo, S. J., G. Homanics, D. J. Schaeffer, L. Schaeffer, J. E. Park, J. Oluoch, T. Zhang, A. Haber, N. T. Seyfried, B. Paten, A. Greenwood, T. Murai, S. H. Choi, H. Huhe, J. Kofler, P. L. Strick, G. W. Carter, and A. C. Silva. 2023. Bridging the rodent to human translational gap: Marmosets as model systems for the study of Alzheimer’s disease. Alzheimer’s & Dementia (N Y) 9(3):e12417.
Sun, J. K., D. Wu, G. C. Wong, T. M. Lau, M. Yang, R. P. Hart, K. M. Kwan, H. Y. E. Chan, and H. M. Chow. 2023. Chronic alcohol metabolism results in DNA repair infidelity and cell cycle-induced senescence in neurons. Aging Cell 22(2):e13772.
Swardfager, W., K. Lanctot, L. Rothenburg, A. Wong, J. Cappell, and N. Herrmann. 2010. A meta-analysis of cytokines in Alzheimer’s disease. Biological Psychiatry 68(10):930-941.
Swerdlow, R. H., and S. M. Khan. 2004. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Medical Hypotheses 63(1):8-20.
Swerdlow, R. H., S. Koppel, I. Weidling, C. Hayley, Y. Ji, and H. M. Wilkins. 2017. Mitochondria, cybrids, aging, and Alzheimer’s disease. In Progress in molecular biology and translational science. Edited by P. H. Reddy. Vol. 146. Cambridge, MA: Academic Press. Pp. 259-302.
Taha, A. 2024. Panelist Remarks. Committee on Research Priorities for Preventing and Treating Alzheimer’s Disease and Related Dementias Public Workshop, Hybrid. January 16–17, 2024.
Tamiz, A. P., W. J. Koroshetz, N. T. Dhruv, and D. A. Jett. 2022. A focus on the neural exposome. Neuron 110(8):1286-1289.
Tejera, D., D. Mercan, J. M. Sanchez-Caro, M. Hanan, D. Greenberg, H. Soreq, E. Latz, D. Golenbock, and M. T. Heneka. 2019. Systemic inflammation impairs microglial Aβ clearance through NLRP3 inflammasome. EMBO Journal 38(17):e101064.
Terron, H. M., S. J. Parikh, S. O. Abdul-Hay, T. Sahara, D. Kang, D. W. Dickson, P. Saftig, F. M. LaFerla, S. Lane, and M. A. Leissring. 2024. Prominent tauopathy and intracellular beta-amyloid accumulation triggered by genetic deletion of cathepsin D: Implications for Alzheimer disease pathogenesis. Alzheimer’s Research & Therapy 16(1):70.
Theurey, P., and P. Pizzo. 2018. The aging mitochondria. Genes (Basel) 9(1):22.
Thurmond, V. A. 2001. The point of triangulation. Journal of Nursing Scholarship 33(3):253-258.
Tijms, B. M., E. M. Vromen, O. Mjaavatten, H. Holstege, L. M. Reus, S. van der Lee, K. E. J. Wesenhagen, L. Lorenzini, L. Vermunt, V. Venkatraghavan, N. Tesi, J. Tomassen, A. den Braber, J. Goossens, E. Vanmechelen, F. Barkhof, Y. A. L. Pijnenburg, W. M. van der Flier, C. E. Teunissen, F. S. Berven, and P. J. Visser. 2024. Cerebrospinal fluid proteomics in patients with Alzheimer’s disease reveals five molecular subtypes with distinct genetic risk profiles. Nature Aging 4(1):33-47.
Traxler, L., J. R. Herdy, D. Stefanoni, S. Eichhorner, S. Pelucchi, A. Szucs, A. Santagostino, Y. Kim, R. K. Agarwal, J. C. M. Schlachetzki, C. K. Glass, J. Lagerwall, D. Galasko, F. H. Gage, A. D’Alessandro, and J. Mertens. 2022. Warburg-like metabolic transformation underlies neuronal degeneration in sporadic Alzheimer’s disease. Cell Metabolism 34(9):1248-1263, e1246.
Traxler, L., R. Lucciola, J. R. Herdy, J. R. Jones, J. Mertens, and F. H. Gage. 2023. Neural cell state shifts and fate loss in ageing and age-related diseases. Nature Reviews Neurology 19(7):434-443.
TREAT-AD (TaRget Enablement to Accelerate Therapy Development for Alzheimer’s Disease). 2024. Two centers, one mission. https://treatad.org/about/ (accessed October 18, 2024).
Trotter, M. B., T. D. Stephens, J. P. McGrath, and M. L. Steinhilb. 2017. The Drosophila model system to study tau action. Methods in Cell Biology 141:259-286.
Uemura, M. T., T. Maki, M. Ihara, V. M. Y. Lee, and J. Q. Trojanowski. 2020. Brain microvascular pericytes in vascular cognitive impairment and dementia. Frontiers in Aging Neuroscience 12:80.
Van Deerlin, V. M., P. M. Sleiman, M. Martinez-Lage, A. Chen-Plotkin, L. S. Wang, N. R. Graff-Radford, D. W. Dickson, R. Rademakers, B. F. Boeve, M. Grossman, S. E. Arnold, D. M. Mann, S. M. Pickering-Brown, H. Seelaar, P. Heutink, J. C. van Swieten, J. R. Murrell, B. Ghetti, S. Spina, J. Grafman, J. Hodges, M. G. Spillantini, S. Gilman, A. P. Lieberman, J. A. Kaye, R. L. Woltjer, E. H. Bigio, M. Mesulam, S. Al-Sarraj, C. Troakes, R. N. Rosenberg, C. L. White, 3rd, I. Ferrer, A. Llado, M. Neumann, H. A. Kretzschmar, C. M. Hulette, K. A. Welsh-Bohmer, B. L. Miller, A. Alzualde, A. Lopez de Munain, A. C. McKee, M. Gearing, A. I. Levey, J. J. Lah, J. Hardy, J. D. Rohrer, T. Lashley, I. R. Mackenzie, H. H. Feldman, R. L. Hamilton, S. T. Dekosky, J. van der Zee, S. Kumar-Singh, C. Van Broeckhoven, R. Mayeux, J. P. Vonsattel, J. C. Troncoso, J. J. Kril, J. B. Kwok, G. M. Halliday, T. D. Bird, P. G. Ince, P. J. Shaw, N. J. Cairns, J. C. Morris, C. A. McLean, C. DeCarli, W. G. Ellis, S. H. Freeman, M. P. Frosch, J. H. Growdon, D. P. Perl, M. Sano, D. A. Bennett, J. A. Schneider, T. G. Beach, E. M. Reiman, B. K. Woodruff, J. Cummings, H. V. Vinters, C. A. Miller, H. C. Chui, I. Alafuzoff, P. Hartikainen, D. Seilhean, D. Galasko, E. Masliah, C. W. Cotman, M. T. Tunon, M. C. Martinez, D. G. Munoz, S. L. Carroll, D. Marson, P. F. Riederer, N. Bogdanovic, G. D. Schellenberg, H. Hakonarson, J. Q. Trojanowski, and V. M. Lee. 2010. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nature Genetics 42(3):234-239.
van Oijen, M., J. C. Witteman, A. Hofman, P. J. Koudstaal, and M. M. Breteler. 2005. Fibrinogen is associated with an increased risk of Alzheimer’s disease and vascular dementia. Stroke 36(12):2637-2641.
Vasic, V., K. Barth, and M. H. H. Schmidt. 2019. Neurodegeneration and neuro-regeneration—Alzheimer’s disease and stem cell therapy. International Journal of Molecular Sciences 20(17):4272.
Vandereyken, K., A. Sifrim, B. Thienpont, and T. Voet. 2023. Methods and applications for single-cell and spatial multi-omics. Nature Reviews Genetics 24(8):494-515.
Vermeulen, R., E. L. Schymanski, A. L. Barabasi, and G. W. Miller. 2020. The exposome and health: Where chemistry meets biology. Science 367(6476):392-396.
Vestin, E., G. Bostrom, J. Olsson, F. Elgh, L. Lind, L. Kilander, H. Lovheim, and B. Weidung. 2024. Herpes simplex viral infection doubles the risk of dementia in a contemporary cohort of older adults: A prospective study. Journal of Alzheimer’s Disease 97(4):1841-1850.
Viggars, A. P., S. B. Wharton, J. E. Simpson, F. E. Matthews, C. Brayne, G. M. Savva, C. Garwood, D. Drew, P. J. Shaw, and P. G. Ince. 2011. Alterations in the blood brain barrier in ageing cerebral cortex in relationship to Alzheimer-type pathology: A study in the MRC-CFAS population neuropathology cohort. Neuroscience Letters 505(1):25-30.
Vineis, P., O. Robinson, M. Chadeau-Hyam, A. Dehghan, I. Mudway, and S. Dagnino. 2020. What is new in the exposome? Environment International 143:105887.
Walsemann, K., N. Hair, M. Farina, P. Tyagi, H. Jackson, and J. Ailshire. 2023. State-level desegregation in the U.S. South and mid-life cognitive function among Black and White adults. Social Science and Medicine 338(116319).
Wang, Z. and G. Chen. 2023. Immune regulation in neurovascular units after traumatic brain injury. Neurobiology of Disease 179:106060.
Wang, X., H. J. He, X. Xiong, S. Zhou, W. W. Wang, L. Feng, R. Han, and C. L. Xie. 2021. NAD+ in Alzheimer’s disease: Molecular mechanisms and systematic therapeutic evidence obtained in vivo. Frontiers in Cell and Developmental Biology 9:668491.
Wang, C., S. Zong, X. Cui, X. Wang, S. Wu, L. Wang, Y. Liu, and Z. Lu. 2023. The effects of microglia-associated neuroinflammation on Alzheimer’s disease. Frontiers of Immunology 14:1117172.
Ward-Caviness, C. K., J. C. Nwanaji-Enwerem, K. Wolf, S. Wahl, E. Colicino, L. Trevisi, I. Kloog, A. C. Just, P. Vokonas, J. Cyrys, C. Gieger, J. Schwartz, A. A. Baccarelli, A. Schneider, and A. Peters. 2016. Long-term exposure to air pollution is associated with biological aging. Oncotarget 15(7):74510-74525.
Watanabe, C., T. Imaizumi, H. Kawai, K. Suda, Y. Honma, M. Ichihashi, M. Ema, and K.-i. Mizutani. 2020. Aging of the vascular system and neural diseases. Frontiers in Aging Neuroscience 12:557384.
WHO (World Health Organization). 2022. Ageing and health. https://www.who.int/news-room/fact-sheets/detail/ageing-and-health (accessed August 16, 2024).
Wild, C. P. 2005. Complementing the genome with an “exposome”: The outstanding challenge of environmental exposure measurement in molecular epidemiology. Cancer Epidemiology, Biomarkers and Prevention 14(8):1847-1850.
Wilker, E. H., M. Osman, and M. G. Weisskopf. 2023. Ambient air pollution and clinical dementia: Systematic review and meta-analysis. BMJ 381:e071620.
Wilkins, H. M., and R. H. Swerdlow. 2021. Mitochondrial links between brain aging and Alzheimer’s disease. Translational Neurodegeneration 10(1):33.
Wilson, R. S., J. A. Schneider, J. L. Bienias, S. E. Arnold, D. A. Evans, and D. A. Bennett. 2003. Depressive symptoms, clinical AD, and cortical plaques and tangles in older persons. Neurology 61(8):1102-1107.
Wilson, R., S. Arnold, J. Schneider, J. Kelly, Y. Tang, and D. A. Bennett. 2006. Chronic psychological distress and risk of Alzheimer’s disease in old age. Neuroepidemiology 27(3):143-153.
Wilson, R. S., S. E. Arnold, J. A. Schneider, Y. Li, and D. A. Bennett. 2007a. Chronic distress, age-related neuropathology, and late-life dementia. Psychosomatic Medicine 69(1):47-53.
Wilson, R. S., K. Krueger, S. Arnold, J. Schneider, J. Kelly, L. Barnes, Y. Tang, and D. Bennet. 2007b. Loneliness and risk of Alzheimer’s disease. Archives of General Psychiatry 64(2):234-240.
Xu, P., M. Wang, W. M. Song, Q. Wang, G. C. Yuan, P. H. Sudmant, H. Zare, Z. Tu, M. E. Orr, and B. Zhang. 2022. The landscape of human tissue and cell type specific expression and co-regulation of senescence genes. Molecular Neurodegeneration 17(1):5.
Yamanaka, S. 2012. Induced pluripotent stem cells: Past, present, and future. Cell Stem Cell 10(6):678-684.
Yan, X., Y. Hu, B. Wang, S. Wang, and X. Zhang. 2020. Metabolic dysregulation contributes to the progression of Alzheimer’s disease. Frontiers in Neuroscience 14:530219.
Yang, W., X. Chen, S. Li, and X. J. Li. 2021. Genetically modified large animal models for investigating neurodegenerative diseases. Cell & Bioscience 11(1):218.
Yin, F., R. Banerjee, B. Thomas, P. Zhou, L. Qian, T. Jia, X. Ma, Y. Ma, C. Iadecola, M. F. Beal, C. Nathan, and A. Ding. 2010. Exaggerated inflammation, impaired host defense, and neuropathology in progranulin-deficient mice. Journal of Experimental Medicine 207(1):117-128.
Yousefzadeh, M. J., J. E. Wilkinson, B. Hughes, N. Gadela, W. C. Ladiges, N. Vo, L. J. Niedernhofer, D. M. Huffman, and P. D. Robbins. 2020. Heterochronic parabiosis regulates the extent of cellular senescence in multiple tissues. GeroScience 42(3):951-961.
Yousefzadeh, M. J., R. R. Flores, Y. Zhu, Z. C. Schmiechen, R. W. Brooks, C. E. Trussoni, Y. Cui, L. Angelini, K. A. Lee, S. J. McGowan, A. L. Burrack, D. Wang, Q. Dong, A. Lu, T. Sano, R. D. O’Kelly, C. A. McGuckian, J. I. Kato, M. P. Bank, E. A. Wade, S. P. S. Pillai, J. Klug, W. C. Ladiges, C. E. Burd, S. E. Lewis, N. F. LaRusso, N. V. Vo, Y. Wang, E. E. Kelley, J. Huard, I. M. Stromnes, P. D. Robbins, and L. J. Niedernhofer. 2021. An aged immune system drives senescence and ageing of solid organs. Nature 594(7861):100-105.
Yu, L., L. B. Chibnik, G. P. Srivastava, N. Pochet, J. Yang, J. Xu, J. Kozubek, N. Obholzer, S. E. Leurgans, J. A. Schneider, A. Meissner, P. L. De Jager, and D. A. Bennett. 2015. Association of brain DNA methylation in SORL1, ABCA7, HLA-DRB5, SLC24A4, and BIN1 with pathological diagnosis of Alzheimer’s disease. JAMA Neurology 72(1):15-24.
Zammit, A. R., D. A. Bennett, and A. S. Buchman. 2023. From theory to practice: Translating the concept of cognitive resilience to novel therapeutic targets that maintain cognition in aging adults. Frontiers in Aging and Neuroscience 15:1303912.
Zhang, B., and V. N. Gladyshev. 2020. How can aging be reversed? Exploring rejuvenation from a damage-based perspective. Advances in Genetics (Hoboken) 1(1):e10025.
Zhang, B., C. Gaiteri, L. G. Bodea, Z. Wang, J. McElwee, A. A. Podtelezhnikov, C. Zhang, T. Xie, L. Tran, R. Dobrin, E. Fluder, B. Clurman, S. Melquist, M. Narayanan, C. Suver, H. Shah, M. Mahajan, T. Gillis, J. Mysore, M. E. MacDonald, J. R. Lamb, D. A. Bennett, C. Molony, D. J. Stone, V. Gudnason, A. J. Myers, E. E. Schadt, H. Neumann, J. Zhu, and V. Emilsson. 2013. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 153(3):707-720.
Zhang, B., J. Weuve, K. M. Langa, J. D’Souza, A. Szpiro, J. Faul, C. Mendes de Leon, J. Gao, J. D. Kaufman, L. Sheppard, J. Lee, L. C. Kobayashi, R. Hirth, and S. D. Adar. 2023a. Comparison of particulate air pollution from different emission sources and incident dementia in the U.S. JAMA Internal Medicine 183(10):1080-1089.
Zhang, L., T. M. Flagan, S. Häkkinen, S. A. Chu, J. A. Brown, A. J. Lee, L. Pasquini, M. L. Mandelli, M. L. Gorno-Tempini, V. E. Sturm, J. S. Yokoyama, B. S. Appleby, Y. Cobigo, B. C. Dickerson, K. Domoto-Reilly, D. H. Geschwind, N. Ghoshal, N. R. Graff-Radford, M. Grossman, G. R. Hsiung, E. D. Huey, K. Kantarci, A. Lario Lago, I. Litvan, I. R. Mackenzie, M. F. Mendez, C. U. Onyike, E. M. Ramos, E. D. Roberson, M. C. Tartaglia, A. W. Toga, S. Weintraub, Z. K. Wszolek, L. K. Forsberg, H. W. Heuer, B. F. Boeve, A. L. Boxer, H. J. Rosen, B. L. Miller, W. W. Seeley, S. E. Lee and ARTFL/LEFFTDS/ALLFTD Consortia. 2023b. Network Connectivity Alterations across the MAPT Mutation Clinical Spectrum. Annals of Neurology 94(4):632-646.
Zhang, L., F. Xu, Y. Yang, L. Yang, Q. Wu, H. Sun, Z. An, J. Li, H. Wu, J. Song, and W. Wu. 2024. PM2.5 exposure upregulates pro-inflammatory protein expression in human microglial cells via oxidant stress and TLR4/NF-κB pathway. Ecotoxicology and Environmental Safety 277:116386.
Zhang, M., and Z. Bian. 2021. Alzheimer’s disease and microRNA-132: A widespread pathological factor and potential therapeutic target. Frontiers in Neuroscience 15:687973.
Zhang, M., A. B. Ganz, S. Rohde, A. J. M. Rozemuller, N. B. Bank, M. J. T. Reinders, P. Scheltens, M. Hulsman, J. J. M. Hoozemans, and H. Holstege. 2023e. Resilience and resistance to the accumulation of amyloid plaques and neurofibrillary tangles in centenarians: An age-continuous perspective. Alzheimer‘s & Dementia 19(7):2831-2841.
Zhang, P., Y. Kishimoto, I. Grammatikakis, K. Gottimukkala, R. G. Cutler, S. Zhang, K. Abdelmohsen, V. A. Bohr, J. Misra Sen, M. Gorospe, and M. P. Mattson. 2019. Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nature Neuroscience 22(5):719-728.
Zhang, W., D. Xiao, Q. Mao, and H. Xia. 2023c. Role of neuroinflammation in neurodegeneration development. Signal Transduction and Targeted Therapy 8(1):267.
Zhang, Z. Y., D. S. Harischandra, R. Wang, S. Ghaisas, J. Y. Zhao, T. P. McMonagle, G. Zhu, K. D. Lacuarta, J. Song, J. Q. Trojanowski, H. Xu, V. M. Lee, and X. Yang. 2023d. TRIM11 protects against tauopathies and is down-regulated in Alzheimer’s disease. Science 381(6656):eadd6696.
Zheng, Y., L. Bonfili, T. Wei, and A. M. Eleuteri. 2023. Understanding the gut–brain axis and its therapeutic implications for neurodegenerative disorders. Nutrients 15(21):4631.





















































































