Charting a Future for Sequencing RNA and Its Modifications: A New Era for Biology and Medicine (2024)
Chapter: 3 Current and Emerging Tools and Technologies for Studying RNA Modifications
3
Current and Emerging Tools and Technologies for Studying RNA Modifications
To understand more fully the role of RNA and its modifications in living systems, researchers will need robust, reproducible, and accessible tools and techniques capable of identifying all RNA modifications, determining their stoichiometry, and elucidating crosstalk (i.e., dependencies) among modification sites. Ideally, this would be accomplished end to end for each RNA molecule in a sample, in a single experiment, and eventually at the single-cell level (Kadumuri and Janga, 2018). Although methods currently exist for measuring global modification levels, and other technologies are in use for sequencing RNA, including some of its modifications, multiple technical limitations and experimental challenges hamper researchers from accomplishing the grander goals listed above. Achieving these goals will require technological advances in the development of reagents, instruments, tools, and technologies for studying RNA modifications.
This chapter presents a summary of available techniques for identifying, quantifying, and sequencing RNA and its modifications, along with current computational and modeling tools used to analyze results. In addition, the chapter identifies technical limitations and challenges that researchers will have to overcome to understand fully the complex and diverse roles of RNA and its modifications in living systems. The chapter closes with some emerging opportunities to advance technologies that could offer new avenues for RNA sequencing endeavors.
CURRENT APPROACHES FOR STUDYING RNA MODIFICATIONS
The presence of modified nucleotides in RNA has been recognized since shortly after RNA was discovered (Davis and Allen, 1957). Complete digestion of different species of RNA, such as transfer RNA (tRNA), ribosomal RNA (rRNA), or messenger RNA (mRNA), to mononucleotides or mononucleosides, followed by thin-layer chromatography or high-performance liquid chromatography–tandem mass spectrometry (LC-MS/MS), revealed the identity and levels of a variety of modifications (Kellner, Burhenne, and Helm, 2010). LC-MS/MS is now the most widely used platform for measuring global modification levels from cells and tissues from different organisms. Any
novel modification can be captured and characterized using these approaches. However, by digesting RNA to mononucleotides or mononucleosides, the sequence context of modifications is lost.
To identify the location and levels of RNA modification at precise coordinates, indirect sequencing approaches1 tailored for measuring individual modifications have been developed. Although informative, these indirect sequencing techniques suffer from several limitations. For example, only a minor portion of the more than 170 modifications are amenable to these currently available approaches (as seen in Table 3-1). Further, in most cases it is not possible to monitor multiple types of modifications in a single experiment using these indirect sequencing techniques, and thus they cannot reveal information about crosstalk between modifications. Finally, indirect sequencing approaches typically require considerable input material and are therefore difficult to adapt to single-cell analysis, which requires very high sensitivity.
As an alternative, direct sequencing2 of RNA molecules—either using mass spectrometry or dedicated methods, such as nanopore sequencing—to reveal the entire RNA sequence, including modifications, hold promise for solving these issues in the near to medium term, once problems such as sequencing length, basecalling, and precision are solved.
Global Modification Measurement
Global modification measurement refers to the process of digesting an RNA sample into its monomer components, which are then separated, analyzed, and identified. In almost all cases, the monomer components are mononucleosides (base + ribose sugar) or mononucleotides (base + ribose sugar + phosphate) (3-1A). Nucleases are most often used for digesting RNA into its monomers. For methods that implement two-dimensional thin-layer chromatography (2D-TLC), digested RNA is typically radiolabeled with phosphorous-32 to permit detection and quantification of the modified nucleotides following the separation step. Then, 2D-TLC is used to separate the different nucleotides and modified nucleotides based on the rates at which they move through the cellulose-coated milieu of the chromatography plates bathed one at a time in two solvents (Grosjean, Keith, and Droogmans, 2004). 2D-TLC reference maps have been compiled for several solvent systems, and these can be used to confirm the identity of the modified mononucleotide (Grosjean, Keith, and Droogmans, 2004; Gupta and Randerath, 1979; Kellner, Burhenne, and Helm, 2010; Stanley and Vassilenko, 1978). When the identity of a modified nucleotide is uncertain, an authentic standard is run on the chromatography plate alongside the sample. Comigration of the standard and a spot from the sample is often the determinant of a positive identification. Modification levels can be quantified based on the level of radioactivity measured upon X-ray film exposure or by phosphor imaging.
While 2D-TLC is fairly simple to implement, the use of radioactive materials is a drawback. A more significant problem is that modifications are identified through inference—that is, by comparing the final migration location on the plate of a sample spot to prior, known mononucleotide migration locations. For those reasons, 2D-TLC has been supplanted by LC-MS/MS, which has become the gold standard for the detection, characterization, and quantification of modified nucleosides (Deng et al., 2022; Heiss et al., 2021; Jora et al., 2019 ; Kellner, Burhenne, and Helm, 2010; Sarkar et al., 2021; Thüring et al., 2016).
The general approach when using LC-MS/MS involves digesting RNA into its constituent nucleosides via enzymatic hydrolysis (Figure 3-1A). As shown in Figure 3-1B, the nucleosides are next subjected to high-performance liquid chromatography (HPLC), which separates the molecules based on their polarity. As the separated nucleosides exit the chromatography column, they are
___________________
1Indirect sequencing refers to methods for sequencing RNA and its modifications that require pretreatment of the RNA with enzymes or chemicals, or conversion to complementary DNA, prior to sequencing.
2Direct sequencing refers to methods for generating information about an RNA’s sequence and modifications by direct analysis of the RNA.
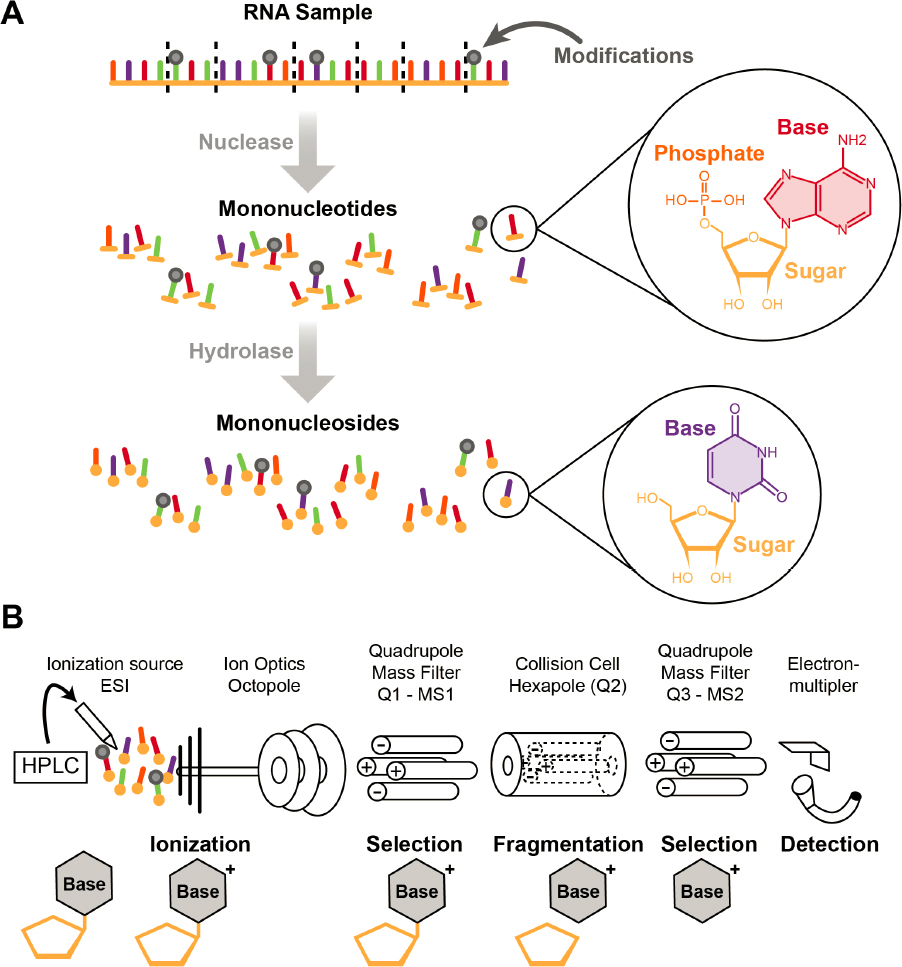
NOTES: (A) The digestion of RNA into mononucleotides (which can be postlabeled for two-dimensional thin-layer chromatography) and mononucleosides. (B) Overview of LC-MS/MS for global analysis of modified nucleosides. A mixture of nucleosides (both modified and unmodified) is separated by high-performance liquid chromatography (HPLC), ionized by electrospray ionization (ESI), and then detected by mass spectrometry. Charged modified nucleosides can be selected and fragmented. Those products, typically the charged nucleobases, are then detected.
SOURCE: Yoluç, 2021. Licensed under Creative Commons Attribution-NonCommercial-NoDerivatives License.
converted to gas-phase ions via electrospray ionization. These gas-phase ions are separated in a mass spectrometer based on their mass-to-charge (m/z) ratio. In tandem mass spectrometry (MS/MS), a particular m/z value is isolated and then dissociated (or fragmented), often via collisions with a neutral inert gas, to generate characteristic product (fragment) ions that can be used to define the molecular structure of the modified nucleoside.
Modified nucleosides are usually identified by three characteristics: the m/z value of the nucleoside, the m/z value(s) of any product ions generated during MS/MS, and the chromatographic retention time. Because nucleoside fragmentation during mass spectrometry typically results in loss of the neutral ribose sugar, the MS/MS step can be used to differentiate between methylations on the nucleobase from those at the 2'-hydroxyl position of the sugar. Moreover, because of this fragmentation behavior, neutral-loss scans can be performed across the entire HPLC elution window to look for m/z values that contain ribose and methylated ribose. These scans provide a routine approach for detecting modified nucleosides. For quantifying modified nucleosides, standard analytical methods can be used (Huang et al., 2023), although calibration curves typically require the presence of modified nucleoside standards, which, as discussed in Chapter 4, are not commercially available for all modified nucleosides.
While modern mass spectrometry platforms are quite sensitive, the amount of sample required for global analysis often depends on the initial complexity of the mixture—that is, how many different RNA species are in the mixture and the abundance of each species. For pure, isolated (single) RNA species, as little as 100 ng of sample can be used; however, if previously unknown modified nucleosides are detected during the analysis, additional sample is required for complete characterization (Clark, Rubakhin, and Sweedler, 2021; Kimura, Dedon, and Waldor, 2020). For more complex mixtures of RNA, global analyses—especially for absolute quantification of modification levels—require µg levels of RNA to start and require the presence of authentic standards for external or internal calibration of the response level of the modification.
One significant problem with LC-MS/MS arises from the process of ionizing the mononucleosides. Typically, electrospray ionization is used, but the effectiveness of this method depends heavily on the polarity of the molecule. More polar molecules are more easily ionized, and less polar molecules generate reduced ion currents. Thus, some modified nucleosides can be ionized and detected readily, even when they are present in the sample at relatively low levels, simply because of their enhanced ionization efficiency. However, less polar molecules may not be detected by this approach unless they are present in significant levels. One approach to addressing this challenge is to derivatize the sample in some fashion to create a more uniform ionization efficiency for all components (Dai et al., 2021; Xie et al., 2022). Another approach is to incorporate a stable isotope, which can then be used to better define the molecular formula that matches the detected precursor and product ions during MS/MS (Cheng et al., 2021; Heiss, Reichle, and Kellner, 2017; Kellner et al., 2014; Wang et al., 2023).
The HPLC step in an LC-MS/MS analysis can also limit the detection of known modifications or discovery of new modifications. Challenges can occur when modified nucleosides co-elute, especially when a modification present at low levels elutes with or close to one of the canonical nucleosides or a high-abundance modification. In such cases, deconvoluting two co-eluting modified nucleosides can be challenging without additional experimental manipulations during the MS/MS step (Janssen et al., 2022; Jora et al., 2018). While modifying the chromatographic conditions may help to resolve the co-eluting nucleosides (Cheng et al., 2021; Lin et al., 2022; Si-Hung, Causon, and Hann, 2017), such changes can result in “new” retention times, necessitating standards for verifying the presence of a particular modification. For these reasons, several investigators have focused on characterizing modifications following separation by alternative methods (Kenderdine et al., 2020) or by developing approaches that do not rely on the chromatographic retention time for identifying the modification (Jora et al., 2022).
Despite the challenges and limitations described above, LC-MS/MS is very effective at routinely providing the complete census of modified RNA nucleosides present in any sample (Kimura, Srisuknimit, and Waldor, 2020; McCown et al., 2020). Moreover, this approach is absolutely necessary when reporting the identity of a new modification (Borek, Reichle, and Kellner, 2020; Dal Magro et al., 2018; Kang et al., 2017; Kimura, Dedon, and Waldor, 2020; Yu et al., 2019).
At the same time, several recent reports document how sample handling and preparation steps prior to global analysis by LC-MS/MS can lead to artifacts that are incorrectly reported as new modifications or can change the chemical structure of the actual modification (Cai et al., 2015; Jora et al., 2021; Kaiser et al., 2021; Matuszewski et al., 2017). These recent reports also demonstrate the need in the field to better define and standardize appropriate sample isolation, preparation, and handling methods for global analyses. Further, while several LC-MS/MS approaches have been reported, the field lacks a set of consensus criteria that must be met before confidently reporting the presence of a new modification. Without such criteria, the field risks spending time and resources following up on artifacts or other experimental errors (Kaiser et al., 2021).
Global RNA modifications measurement can be improved by developing methods that require less sample input, enable higher confidence and accuracy in identifying and quantifying modifications, and make experimental protocols accessible to contract research organizations and core labs; these objectives represent key milestones to achieving the goals of this report.
Indirect Sequencing
Currently, available technology for indirect sequencing methods uses a reverse transcriptase enzyme to transcribe RNA molecules to complementary DNA (cDNA), followed by sequencing of the cDNA. During analysis of the cDNA sequence, the template RNA base sequence (adenine [A], cytosine [C], guanine [G], uracil [U]), and sometimes, additional information about modified nucleotides, is derived. However, many RNA modifications become invisible during the preparation of cDNA, and additional steps need to be taken prior to reverse transcription to reveal the positions of the modifications. In one strategy, RNA fragments containing the modification of interest are isolated using antibodies against the modification prior to cDNA preparation (e.g., RNA immunoprecipitation sequencing, cross-linking and immunoprecipitation sequencing). In a second approach, treatment of the RNA with chemicals or enzymes specifically marks the modified bases so that they can be recognized by reverse transcription and appear as a misincorporation/mutation, deletion, or stop signature in the cDNA (Table 3-1).
The hydrolytic deamination of adenosines in RNA to inosines (A-to-I) by adenosine deaminases that act on RNA (ADAR enzymes) was the first widespread modification identified in eukaryotic mRNA (Reich and Bass, 2019). Because, like guanosine (G), inosine prefers to pair with cytidine (C), inosine can be revealed easily, without any pretreatment, by identifying A-to-G transitions following reverse transcription and sequencing of cDNA (Reich and Bass, 2019).
While it took heroic efforts to detect RNA methylation, including N6-methyladenosine (m6A), in mRNA in the 1970s (Desrosiers, Friderici, and Rottman, 1974), in-depth characterization of m6A became possible once specific m6A antibodies were developed and used for immunoprecipitation of fragmented, m6A-modified RNA followed by next-generation sequencing (NGS) of the cDNA3 (Dominissini et al., 2012; Meyer et al., 2012). In vitro crosslinking of m6A-modified RNA with m6A-specific antibodies further improved the resolution, permitting precise mapping of the modification sites (Ke et al., 2015; Linder et al., 2015). This precision mapping was possible because during the reverse transcription process, crosslinking-induced mutations were detectable in sequencing
___________________
3Next-generation sequencing, often called NGS, refers to several high-throughput methods for sequencing DNA and indirectly RNA (by conversion of RNA to cDNA) (see Chapter 1 and Figure 1-6).
as the sites where modified bases had been in the original mRNA. This antibody approach was adapted to map potential N1-methyladenosine (Dominissini et al., 2016) and N4-acetylcytidine modification sites (Arango et al., 2018); resulting data suggest that both modifications play a role in the regulation of protein synthesis (Roberts, Porman, and Johnson, 2021).
Table 3-1 summarizes a multitude of indirect sequencing methods that incorporate either antibody enrichment or chemical/enzymatic reaction of modified RNA nucleotides, or in a few cases, alternative strategies. In general, modification mapping can involve either positive readout that detects the modification as a mutation, deletion, or stop signature in the sequencing data, or negative readout that detects the unmodified base. Examples include pseudouridine (Ψ) versus 5-methylcytosine (m5C) sequencing using bisulfite treatment, in which Ψ is read out through a deletion signature, m5C is read out as C, and unmodified Cs are read out as thymine (T).
Immunoprecipitation-based methods suffer from specificity issues, most often because the antibody has some degree of cross-reactivity with related nucleosides (Garcia-Campos et al., 2019; Grozhik et al., 2019). In addition, such methods do not allow for a quantitative assessment of modification levels at the identified sites. Alternative approaches exploit differential sensitivity of modified sites to ribonucleases (RNases) (MAZTER-seq, m6A-REF-seq; see Table 3-1); however, this works only for the few cases for which such nucleases exist. Some base modifications, including Ψ (Carlile et al., 2014; Schwartz et al., 2014), N4-acetylcytidine (ac4C) (Sas-Chen et al., 2020), m5C, and m6A (Hu et al., 2022; Liu et al., 2023), can be derivatized chemically or enzymatically to increase specificity (see Table 3-1). Typically, such derivatization can then be revealed after reverse transcription and sequencing, either as sites of stalling for reverse transcriptase or as reverse transcriptase–induced cDNA sequence variants, analogous to the identification of inosines in mRNA by A-to-G transitions in RNA-sequenced datasets (Roth, Levanon, and Eisenberg, 2019).
Indirect sequencing of modified RNA transcripts has several shortcomings. NGS methods yield RNA modification information from mutation, deletion, and stop signatures in reads aligned to reference sequences. These signatures are obtained during cDNA synthesis when the reverse transcriptase enzyme reacts to modified nucleotides, sometimes in unexpected ways. While these methods can identify and even quantify specific modifications at single-base resolution in each transcript, the information obtained is at the bulk-transcript level, not at the single-molecule level; thus, tissue- or cell-specific information is lost. Since most protocols involve an immunoprecipitation or derivatization designed to reveal only one modification, current NGS protocols typically reveal only the modification of interest and do not provide information on the interplay between different RNA modifications.
Third, NGS methods generate short reads of up to 300 nucleotides, which, on average, represent less than 15 percent of a mammalian mRNA transcript (Lander et al., 2001), thus precluding the assessment of long-range coordination of modification sites. The short-read length also complicates calling of splice isoform–specific or single transcript–specific modifications (see Figure 1-6). In addition, unavoidable false-positive calls confound data interpretation for technologies requiring chemical/enzymatic treatment or the use of antibodies, particularly for low-abundance modifications (Kong, Mead, and Fang, 2023). For example, chemical labeling of Ψ by N-cyclohexyl-N′-(2-morpholinoethyl)carbodiimide metho-p-toluenesulphonate (CMC) (Carlile et al., 2014; Schwartz et al., 2014) results in false positives due to nonspecific labeling at sites other than Ψ (Wiener and Schwartz, 2021). While commonly used antibodies typically have hundred- to thousand-fold higher affinity for modified over nonmodified bases, there may be considerable background noise within the enormous sequence space of the transcriptome. As an example, a study using m6A antibodies and nonmethylated, in vitro–transcribed RNA found 10,000–15,000 peaks that could be misinterpreted as interaction sites (Zhang et al., 2021). These limitations of indirect sequencing methods are a strong impetus for the development of direct methods for determining epitranscriptomes.
TABLE 3-1 Summary of Indirect Sequencing Techniques Applied to Specific RNA Modifications
| Modification | Method | Ab-based | Chemical Modification | Computational Readout | Quantitation of Modification Levels | Reference |
|---|---|---|---|---|---|---|
| m6A | m6A-seq | yes | no | Sequence enrichment | no | Dominissini et al., 2012 |
| m6A-seq2 | yes | no | Sequence enrichment | no | Dierks et al., 2021 | |
| m6A-LAICseq | yes | no | Sequence enrichment | yes | Molinie et al., 2016 | |
| GLORI | no | yes | RT–induced variant at nonmethylated A | yes | Liu et al., 2023 | |
| m6A-SAC-seq | no | yes | RT-induced random variant at m6A | yes | Hu et al., 2022 | |
| DART-seq | no | no | C-to-U variants close to m6A sites induced by expression of YTH-APOBEC1 fusion | no | Meyer, 2019 | |
| MAZTER-seq | no | no | Inference of m6A levels from cleavage activity of a m6A sensitive RNase | yes co | Garcia-Campos et al., 2019 ntinued | |
| m6A-REF-seq | no | no | Inference of m6A levels from cleavage activity of a m6A sensitive RNase | yes | Zhang et al., 2019 | |
| scDART-seq | no | no | C-to-U variants close to m6A sites induced by expression of YTH-APOBEC1 fusion | no | Tegowski, Flamand, and Meyer, 2022 | |
| miCLIP | yes | no | Crosslinkinginduced RT-stall | no | Linder et al., 2015 | |
| meCLIP | yes | no | Crosslinkinginduced RT-stall | no | Roberts, Porman, and Johnson, 2021 | |
| m6A-CLIP | yes | no | Sequence enrichment | no | Ke et al., 2015 | |
| m6A-SEAL-seq | no | yes | Sequence enrichment | no | Wang et al., 2020 | |
| eTAM-seq | no | yes | Enzymatic A-to-I variants at nonmethylated A | yes | Xiao et al., 2023 |
| Modification | Method | Ab-based | Chemical Modification | Computational Readout | Quantitation of Modification Levels | Reference |
|---|---|---|---|---|---|---|
| m1A | m1A-seq | yes | no | Sequence enrichment | no | Dominissini et al., 2016) |
| ac4C | acRIP-seq | yes | no | Sequence enrichment | no | Arango et al., 2018 |
| ac4C-seq | no | yes | RT-induced random variant at chemically modified ac4C | yes | Sas-Chen et al., 2020 | |
| m3C | HAC-seq | no | yes | Mapping of C insensitive to hydrazine/aniline cleavage | yes | Cui et al., 2021 |
| m7G | MeRIP-seq | yes | no | Sequence enrichment | Lin et al., 2018 | |
| TRAC-seq | yes | yes | Insensitivity of m7G to NaBH4 reduction is exploited | Lin et al., 2018 | ||
| BoRed-seq | no | yes | Insensitivity of m7G to NaBH4 reduction is exploited | Pandolfini et al., 2019 | ||
| Ψ | CeU-seq | no | yes | Biotin is conjugated to N3-CMC-Ψ through click chemistry | Li et al., 2015 | |
| PRAISE | no | yes | Bisulfite-induced deletion signature during reverse transcription | Zhang et al., 2023 | ||
| Pseudo-seq | no | yes | Crosslinkinginduced RT-stall at CMC-modified sites | no | Carlile et al., 2014 | |
| Pseudo-seq | no | yes | Crosslinkinginduced RT-stall at CMC-modified sites | no | Schwartz et al., 2014) | |
| HydraPsiSeq | no | yes | Mapping of uridines insensitive to hydrazine/aniline mediated cleavage | yes | Marchand et al., 2020 | |
| BID-seq | no | yes | Chemical-induced RT skipping at Ψ sites read as deletion | yes | Dai et al., 2023) |
| Modification | Method | Ab-based | Chemical Modification | Computational Readout | Quantitation of Modification Levels | Reference |
|---|---|---|---|---|---|---|
| Inosine | AEI | no | no | A-to-G variants in RNA-seq | yes | Roth, Levanon, and Eisenberg, 2019 |
| ICE-seq | no | yes | A-to-G variants in RNA-seq | yes | Okada et al., 2019 | |
| m5C | BS-seq | no | yes | Specific variant in bisulfite-treated RNA | yes | Schaefer et al., 2009 |
| m5C-RIP-seq | yes | no | Sequence enrichment | no | Xue, Zhao, and Li, 2020 | |
| Nm | RibOxi-Seq | no | yes | Mapping of periodateinsensitive RNA 3’ends after random fragmentation | no | Zhu, Pirnie, and Carmichael, 2017 |
| RiboMethSeq | no | no | Mapping of 3’ ends protected from hydrolysis by Nm | yes | Marchand et al., 2016 | |
| Nm-mut-seq | no | no | cDNA synthesis using an evolved RT generates variants at Nm | yes | Chen et al., 2023 | |
| Abasic sites | APE1 RIP | yes | no | Sequence enrichment | no | Liu et al., 2020 |
| s4U | TUC-seq | no | yes | T-to-C conversion induced by OsO4 | yes | Riml et al., 2017 |
| SLAM-seq | no | yes | Alkylation of s4U, resulting in T-to-C variants in cDNA | yes | Herzog et al., 2017 | |
| TimeLapse-seq | no | yes | Converts s4U into C | yes | Schofield et al., 2018 | |
| m5C, Ψ, m1A | RBS-seq | no | yes | Bisulfite converts C-to-T, Ψ to deletion, m1A by mutation signature | Khoddami et al., 2019 | |
| m1A, m1I, m1G, m3C, m22G, m3U | DM-tRNA-seq | no | no | Mutation signature validated by demethylase reversal | Clark et al., 2016 |
NOTE: CMC = N-cyclohexyl-N′-(2-morpholinoethyl)carbodiimide metho-p-toluenesulphonate. Other abbreviations are defined in the Front Matter.
Single-Molecule and Direct Sequencing Technologies
Third-Generation Sequencing
Unlike NGS methods, which require DNA amplification and have short-read lengths, third-generation sequencing (TGS) methods can sequence single molecules at read lengths that allow sequencing of very long DNA molecules, or a complete RNA molecule, even for very long mRNAs. In theory, some TGS methods have the potential to produce unique signatures for each RNA modification, although the technology for this has not yet been developed. Therefore, TGS offers the potential for simultaneous identification of all modification sites and types in a single RNA molecule of any length, from end to end. Currently, two TGS methods are widely available commercially: Oxford Nanopore Technologies4 (ONT) and Pacific Biosciences24F5 (PacBio).
PacBio uses zero-mode waveguide wells that allow imaging of nucleic acid synthesis processes—a “sequencing-by-synthesis” method. When used for DNA sequencing, each well contains an immobilized DNA polymerase enzyme that binds a single-primer template DNA molecule. The nucleotides that are added for synthesis are each labeled with a different fluorophore. Sequencing is accomplished by capturing and recording the residual time of fluorescent substrates used for synthesis, which is characteristic for each nucleotide. In its current form, this method is indirect for RNA sequencing, as it requires prior conversion to cDNA by reverse transcription. Nevertheless, by substituting the immobilized DNA polymerase with a suitable reverse transcriptase, the PacBio method could, in principle, be used for single-molecule direct RNA sequencing. Although a PacBio method for RNA sequencing without cDNA conversion has yet to be applied commercially (possibly because of the difficulty of engineering highly processive reverse transcriptases suitable for the PacBio platform), the principle of applicability was published in 2013 to study m6A in mRNA (Vilfan et al., 2013). In this study m6A in mRNA was identified by a much longer dwell time between its cDNA synthesis and adjacent residues. The same principle was used for PacBio sequencing of N6-methyldeoxyadenosine in genomic DNA.
Nanopore sequencing represents a direct RNA sequencing method, meaning the RNA and their modifications are measured directly, rather than in a cDNA made from the RNA (as in NGS methods or in the PacBio method). ONT uses an array of protein pores embedded in a synthetic membrane barrier, across which a current is applied. Protein helicase capture of DNA or RNA molecules attached to specific adaptors allows for single-stranded nucleic acid molecules to move through the pore end to end, producing a current signature (called squiggles) that optimally enables bases, including modified bases, to be read (Stoddart, et al., 2010a,b). ONT suffered early on from high error rates, but thanks to advances in nanopore chemistry and basecalling algorithms, the error rates for nanopore RNA sequencing have been reduced significantly, to less than 5 percent (Wang et al., 2021). A substantial portion of the remaining errors were due to modifications to native sequences of DNA and RNA (Wick et al., 2019). See Box 3-1 for additional technical details of nanopore sequencing.
Since 2018, ONT-based sequencing has been widely applied to the study of mRNA modifications, primarily looking for the most abundant mammalian mRNA modifications: m6A, Ψ, and inosine. For example, one study used ONT’s technology to sequence a transcriptome of human polyadenylated mRNA from a cultured cell line, identified numerous isoforms, and compared a specific mRNA in vitro transcript and its cellular counterpart to show differences in the current between modified and unmodified sites, including m6A and A-to-I modifications (Workman et al., 2019). In another study, ONT analysis found that m6A modifications can appear as errors in nanopore sequencing. The researchers used a difference in the percent error of specific bases to
___________________
4See https://nanoporetech.com/ (accessed October 27, 2023).
5See https://www.pacb.com/ (accessed October 27, 2023).
BOX 3-1
Nanopore Sequencing
Oxford Nanopore Technologies (ONT) has developed real-time sequencing devices capable of producing 50–250 gigabases of DNA and RNA sequencing data. Their instruments, ranging from benchtop to pocket size, are capable of direct RNA sequencing (i.e., do not require polymerase chain reaction [PCR] amplification and complementary DNA [cDNA] prep), although the types of modifications they can detect are currently limited. For example, the MinION is a compact and inexpensive sequencing instrument that has been used for target-specific sequencing of the influenza A viral genome and human DNA (Gilpatrick et al., 2020; Keller et al., 2018). When sequencing polyadenine (polyA) messenger RNA (mRNA), an oligo-deoxythymine (dT) bead step is included to enrich for the polyA mRNA, which is followed by cDNA strand synthesis using an oligo-dT primer (Figure 3-2A). The RNA–DNA molecule created during cDNA synthesis prevents RNA secondary structures from forming and protects the RNA from degradation by most ribonucleases, leading to increased throughput. Nucleic acids are prepped for nanopore sequencing by ligating an RNA-sequencing adapter to the 3’ end of the cDNA–RNA hybrid molecule. A “motor protein” is attached to the adapter’s 3’ end and a “tether” protein is bound to its 5’ end. Once the complex makes contact with a nanopore, the tether protein prevents the cDNA from passing through the pore, ensuring it is not sequenced. The motor protein, meanwhile, drives the other nucleic acid strand through the nanopore at a rate of approximately 450 bases per second for DNA and approximately 70 bases per second for RNA.
An active area of research is the development of new (synthetic) nanopores and the reengineering of existing ones to increase the lifetime of the nanopores to facilitate improvements in the sequencing throughput while maintaining the accuracy of the detected bases. A number of naturally occurring pores are in use, such as the MspA pore from Mycobacterium smegmatis and the alpha hemolysin pore from Staphylococcus aureus (MacKenzie and Argyropoulos, 2023). ONT utilizes an engineered protein derived from the Escherichia coli CsgG pore, which it embeds in an electrically resistant membrane made from a synthetic polymer (Figure 3-2B) (Ayub and Bayley, 2016; Goyal et al., 2014; Henley, Carson, and Wanunu, 2016; Ip et al., 2015).
The MinION flow cell membrane contains 2,048 individually addressable pores, grouped in 512 channels. MinKNOW™, the first edition of the software provided by ONT for the MinION device, handles various core tasks, including run-parameter assignment, data acquisition, and feedback on the experiment’s progress. MinKNOW assigns four pores per channel, allowing 512 molecules to be sequenced simultaneously (Ip et al., 2015). During sequencing, a sensor measures the current fluctuations, generated by nanopores immersed in an ionic solution, several thousand times per second (Deamer, Akeson, and Branton, 2016; Jain et al., 2016).
Application of a voltage causes an ionic current to pass through the nanopore. As the nucleic acid strand moves through the nanopore from one chamber to the other, the current changes in a characteristic way that allows identification of the nucleic acid sequence. Unlike Sanger sequencing, which recognizes individual nucleotides, nanopore sequencing considers the five or six nucleotides present in the pore to produce a raw signal. These raw signals, represented as “squiggle” plots (Figure 3-4C), provide information about the changes in current from all possible K-mers (six for DNA and five for RNA) occupying the sensor pore at a given time point. For unmodified RNA, this results in 1,024 (45) different signal configurations (Bayley, 2015; Simpson et al., 2017; Wick, Judd, and Holt, 2019; Zorkot, Golestanian, and Bonthuis, 2016). Notably, the signal obtained for a specific sequence may vary between different runs, necessitating the use of signal means from several runs to accurately identify the sequence of a nucleic acid (Figure 3-2D).

NOTES: (A) PolyA+ messenger RNA (mRNA) is first enriched using oligo-dT primers and then ligated to sequencing adapters, which are bound to motor and tether proteins. (B) An array of sequencing nanopores is shown. When the nucleic acid–protein complex contacts a pore, the motor protein helps the RNA molecule pass through the nanopore in a 3’–5’ direction. Bases passing through the nanopore cause a characteristic disruption in the current, which is stored as the raw signal. The inset on the right shows a squiggle plot of the raw signal. (C) The raw signal is converted into bases using a basecalling algorithm. The plot separates normal and modified signals for a GCAGG oligonucleotide (multiple signal traces are shown, with the average highlighted in color). The left panel represents signal traces for an unmodified oligonucleotide context, while the right panel represents signals originating from the corresponding GC(m6A)GG oligonucleotide regions in the transcriptome. Data shown are idealized representative traces; real data often contain significant degrees of noise.
SOURCE: Anreiter et al., 2021, with permission from Elsevier.
detect modification sites based on data from synthetic modified RNA (Jenjaroenpun et al., 2020; Pratanwanich et al., 2021).
A few studies recently developed characteristic basecalling error signatures in the nanopore data for Ψ, which can be used to estimate per-site modification stoichiometry and to detect multiple Ψ sites at single-molecule resolution (Begik et al., 2021; Hassan et al., 2022; Tavakoli et al., 2023). It has been applied to mRNAs and rRNAs in yeast, and following oxidative, cold, and heat stress (see nanoRMS in Table 3-3) (Begik et al., 2021) and on human transcriptomes (Hassan et al., 2022; Tavakoli et al., 2023). Burrows and colleagues (Fleming et al., 2021) used 16S and 23S rRNA strands from Escherichia coli and corresponding synthetic controls in a series of studies in multiple-ribosomal RNA sequence contexts to identify differences in basecalling, ionic current, and dwell time for more than 16 different RNA modification types. They determined that the passage of Ψ through a helicase brake sensor resulted in a pause of the RNA molecule, producing a long-range dwell time, which was used to analyze SARS-CoV-2 nanopore sequencing data. Results supported the presence of five conserved Ψ sites in the viral RNA genome and rejected other sites previously reported (Fleming et al., 2021).
Given the abundance of m6A and Ψ in mRNA, multiple researchers have developed computational tools for model training, modification detection, and quantitation of m6A (Fleming et al., 2021; Gao et al., 2021; Hendra et al., 2022; Leger et al., 2021; Liu, et al., 2019a, 2022; Lorenz et al., 2020) and Ψ (Gao et al., 2021; Hendra et al., 2022; Leger et al., 2021; Liu et al., 2019a, 2022; Lorenz et al., 2020) in mRNA and other RNA types (Hassan et al., 2022; Huang et al., 2021; Li et al., 2021; Ramasamy et al., 2022). Note, however, that a systematic comparison of currently available computational tools used to decode RNA modifications from native RNA sequences found that the tools varied in sensitivity, precision, and bias (Zhong et al., 2023). Thus, while exciting developments have been made, significant improvement and progress are needed in methods such as ONT nanopore sequencing, in order for the goals this report envisions to be achieved.
Mass Spectrometry-based Methods
In addition to global analysis of modified nucleosides, LC-MS/MS has been used for direct sequencing and modification mapping of RNAs, typically more abundant RNAs (e.g., tRNAs, rRNAs). The only effective approach for mass spectrometry is to start with RNAs in which the canonical sequence (A, C, G, U) is known, which is then analyzed strictly using mass spectrometry to place modifications at specific sequence locations. Two general approaches have been developed that take advantage of the unique capabilities of mass spectrometry to directly identify the presence and location of modifications: bottom-up and top-down mapping.
Bottom-up modification mapping. The more common approach is so-called bottom-up modification mapping. As with proteomics (Shuken, 2023), this approach involves digesting intact RNA into smaller oligomers that can be separated by HPLC and sequenced in the gas phase by MS/MS. The RNA is digested by specific nucleases or other chemical treatments (Jora et al., 2019) (Figure 3-3). For example, ribonuclease T1 cleaves RNA on the 3’-side of G residues, leading to digestion products that contain only a single G at the 3’-terminus (Table 3-2). This generates a mixture of oligoribonucleotides that are usually of a length compatible with standard LC-MS/MS instrumentation and its capabilities to perform collision-induced dissociation, referred to by some as collisionally activated dissociation (Figure 3-4).
Separation of the oligonucleotides prior to mass spectrometry analysis was improved dramatically in 1997 with the introduction of an ion-pair reversed-phase chromatography solvent system (Apffel et al., 1997). This system uses two components: an alkylamine effective at enhancing interactions of the oligonucleotide with the HPLC stationary phase and hexafluoroisopropanol that leads to enhanced ionization during electrospray ionization. To date, this general solvent system
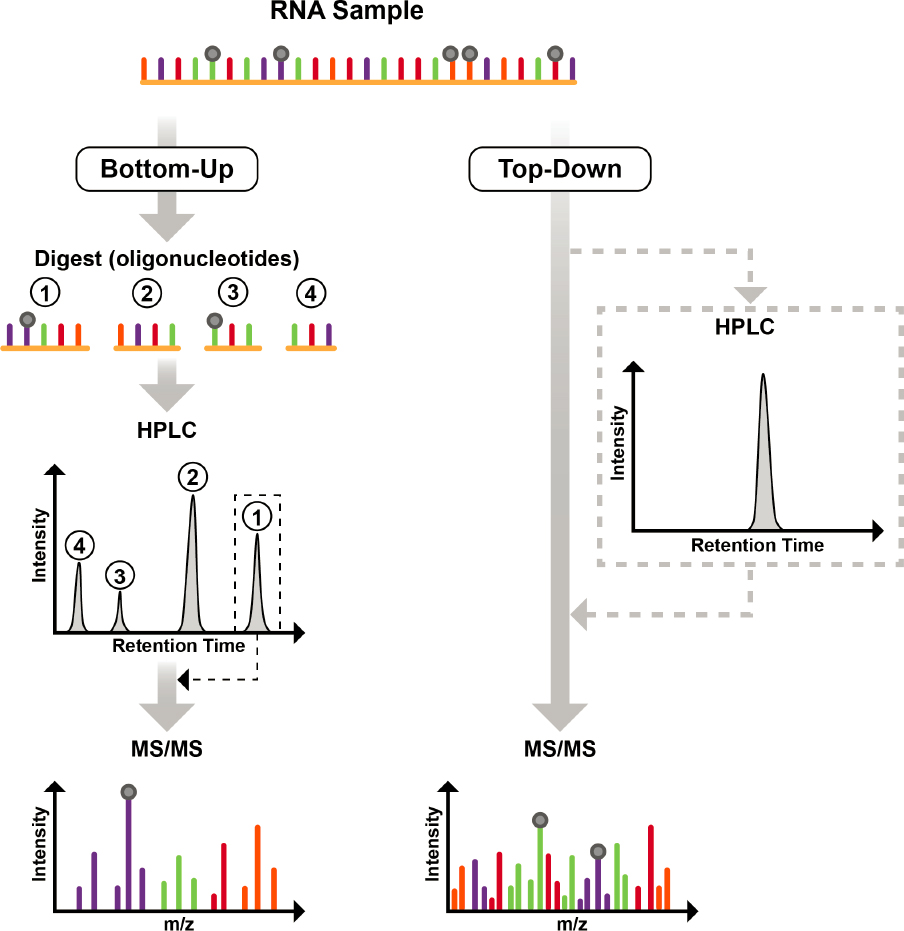
NOTES: In the bottom-up approach (left-hand side), an RNA sample is digested into smaller pieces, oligonucleotides, which are then separated by high-performance liquid chromatography (HPLC) and analyzed online by tandem mass spectrometry (MS/MS) to yield information about sequence and modification location and abundance. In the top-down approach (right-hand side), a pure RNA sample is analyzed by MS/MS to yield information about sequence and modification location and abundance. To generate pure RNAs, mixtures can be separated by offline HPLC, and the RNA of interest can be collected as it elutes from the HPLC at a specific retention time.
TABLE 3-2 Specificity and Limitations of Enzymes Used for RNA and Oligonucleotide Digestion
| Enzyme | Cleavage Specificity | Limitation | Reference |
|---|---|---|---|
| RNase T1 | 3' terminus of unmodified guanosine and the modified nucleoside N2methyl guanosine | RNase T1 does not generate high sequence coverage, especially when G-rich sequence redundancies are available | Greiner-Stöffele, Foerster, and Hahn, 2000 |
| RNase A | 3' terminus of all canonical pyrimidines and pseudouridine | RNase A generates shorter degradation products that are not useful for modification placement | Prats-Ejarque et al., 2019 |
| RNase U2 | 3' terminus of canonical purines with a slight selectivity towards adenosine | RNase U2 does not increase the sequence coverage of mapped modifications | Houser et al., 2015 |
| MC1 | 5' terminus of pseudouridine and unmodified uridines | Commercially unavailable | Addepalli, Lesner, and Limbach, 2015 |
| Cusativin | 3' terminus of unmodified cytidine and the modified nucleoside 5-methylcytidine (m5C) | Commercially unavailable | Addepalli et al., 2017 |
| Human RNase 4 | 3' terminus of uridine residues prior to purines (slight preference for U-A relative to U-G) | Generally insensitive to modified uridines | Wolf et al., 2022 |
NOTE: Abbreviations are defined in the Front Matter.
remains the best for separating oligonucleotides up to 50-mers with high chromatographic resolution (Donegan, Nguyen, and Gilar, 2022). However, because the solvent system is highly corrosive, it requires dedicated hardware, generates carryover effects when switching mass spectrometry instrumentation from negative to positive polarity, and is a health hazard to users. For those reasons, there is ongoing work on alternative solvent systems and stationary phases that can separate complex mixtures of oligonucleotides without the downsides of the current ion-pair solvent system (Demelenne et al., 2020; Hagelskamp et al., 2020).
Because mass spectrometry measures an intrinsic property of any compound (i.e., the molecular weight), regardless of the type of mass spectrometry instrument used, the molecular weight will not vary. Moreover, the more precisely (and accurately) one can measure molecular weight (or the m/z value), the easier it is to define the chemical composition of the molecule. In the early 1990s, it was shown that the number of A, G, C, and U (i.e., the base composition) in an oligonucleotide could be determined simply by using mass spectrometry to measure the molecular weight of the oligonucleotide. This would work if (a) the m/z of the oligonucleotide could be measured with high levels of precision and (b) a constraint was placed on one of the canonicals within the base composition (Pomerantz, Kowalak, and McCloskey, 1993). This key factor is one reason that digestion of RNAs via RNase T1 is preferred—because this nuclease cleaves at all unmodified G residues, any digestion product would contain a single G (assuming no modified Gs are unrecognized by the nuclease). Thus, digesting RNAs with RNase T1 generates a pool of oligonucleotides that all have a known base composition, which simplifies determining the oligonucleotide sequence and modification placement. More importantly, because the measured molecular weight of an RNase T1 digestion product must agree to a known base composition, the presence of modifications in that measured digestion product is immediately discernible because of the mass difference between the measured value and the closest base composition of lower molecular weight. These key insights allowed the expansion of direct sequence placement of modifications onto tRNAs and rRNAs and
even provided a route for using direct sequencing to identity constituent tRNAs within an unseparated mixture (Felden et al., 1998; Guymon et al., 2006; Hossain and Limbach, 2007; Kowalak et al., 1993; Wagner et al., 2004).
The MS/MS step is key in a bottom-up method. Because oligonucleotides have a very robust and reproducible fragmentation pattern along the phosphodiester backbone during collision-induced dissociation, a mass ladder is created whereby the identity and sequence of each nucleotide residue can, in principle, be determined based on the difference in mass between each of the 5'- and 3'-dissociation products of the same type (Figure 3-4) (McLuckey, Van Berkel, and Glish, 1992). For example, the mass difference between the dn and dn+1 fragment ions can be used to define the identity of the nucleotide (Figure 3-4). Moreover, because one only needs to measure the difference in two m/z values to define the identity of the nucleotide, the mass accuracy and resolution requirements during MS/MS can be less restrictive than those during the first stage of mass analysis.
One powerful feature of mass spectrometry–based modification mapping is that all known posttranscriptional modifications, except Ψ (an isomer of U), result in an increase to the mass of the canonical nucleotide. Because mass spectrometry specifically detects m/z values of ions, any posttranscriptional modifications are readily denoted by the unique m/z difference in the mass ladders created during MS/MS. For example, a single methylation to a canonical nucleotide results in a +14-dalton (Da) mass increase, thiolation (the replacement of an oxygen atom with a sulfur atom) results in a +16-Da mass increase, and so on. Thus, using mass spectrometry and MS/MS data,
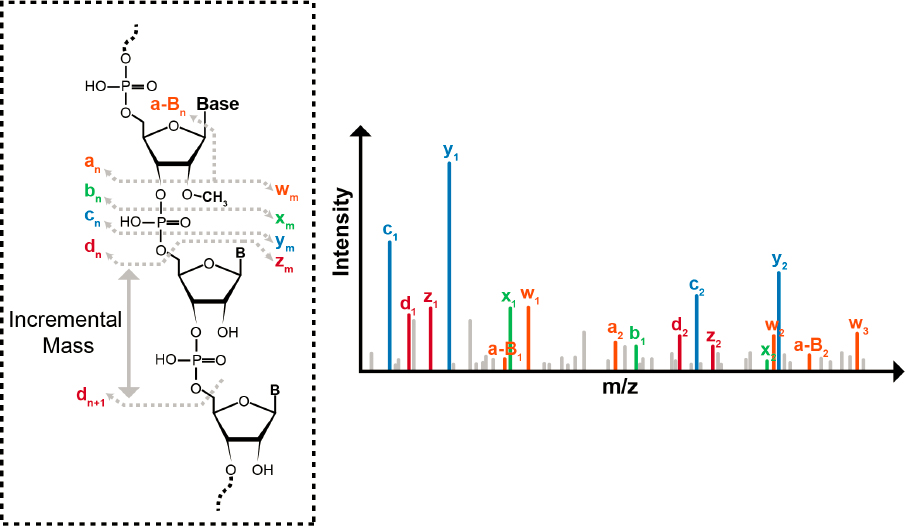
SOURCE: D’Ascenzo, 2022. Licensed under Creative Commons Attribution 4.0 International License.
the sequence can be reconstructed, and the locations of modified nucleosides can be identified by evaluating the mass differences in the various mass ladders generated during analysis.
The bottom-up method was first demonstrated in 1993 (Kowalak et al., 1993). Initial applications using only LC-MS were focused on rRNA, which could be isolated as a homogeneous sample in relatively large amounts (Kowalak, Bruenger, and McCloskey, 1995; Kowalak et al., 1993). The introduction of MS/MS and additional purification strategies enabled modification mapping of other types of RNAs, often tRNAs with their many modifications (Crain et al., 2002; Suzuki and Suzuki, 2014). While these tRNA samples were purified to be homogeneous prior to mapping, more recently, bottom-up modification mapping has been applied to mixtures of tRNAs (Puri et al., 2014; Yu et al., 2019).
Several factors limit the widespread use of mass spectrometry for direct sequencing and modification mapping of RNA.
First, the bottom-up method generates some RNA digestion products that are uninformative because of sequence redundancies. For example, digestion with RNase T1 can generate monomers (single Gp’s), dimers, trimers (e.g., AUGp, AAGp), and so on that appear at multiple sites within the overall RNA sequence. Should one of these digestion products be modified—for example [m6A] UGp—the approach cannot differentiate which GAUG within the RNA contains the modification. One strategy for resolving such ambiguities is to perform multiple digestions, each using a different nuclease (see Table 3-2) to generate sufficient overlap to verify a specific modification site (Thakur et al., 2020).
Second, conventional MS/MS approaches used to generate mass ladders are less reliable for oligomers that are greater than 40 nucleotides long (Hannauer et al., 2023). Because conventional MS/MS approaches deposit a fixed overall amount of energy into the oligomer, longer oligomers have less energy per internucleotide linkage, which can reduce fragmentation (sequencing read lengths), leading to greater uncertainty about the overall sequence and where modifications are located. This is a significant obstacle when conducting de novo sequencing of modified RNAs to discover sites of modification. Thus, the ability to generate longer reads along with higher-quality sequence data from those reads would be a major advance for this method.
Third, mass spectrometry requires microgram or greater amounts of sample (Kimura, Dedon, and Waldor, 2020). Ample digestion, purification for mass spectrometry compatibility, and on-line chromatographic separation are all necessary prior to the sample reaching the mass spectrometer, and each step can lead to sample loss. New and much more sensitive workflows and instrumentation advances are necessary to reduce sampling requirements.
Fourth, very few commercial software packages, either standalone or included with an LC-MS platform, are dedicated to the high-throughput analysis of LC-MS/MS data for mapping modifications onto RNA transcript sequences. Initial programs have been open source (D’Ascenzo et al., 2022; Sample et al., 2015; Wein et al., 2020; Yu et al., 2017), but mass spectrometry vendors are beginning to provide platform-specific software packages tailored for analyzing LC-MS/MS data for the presence of modifications. These packages, however, are designed primarily for analyzing synthetic oligonucleotides, which may have few or no modifications (e.g., small interfering RNAs, guide RNAs). In addition, these programs are geared towards confirming sequencing data against a known (e.g., synthetic) sequence and do not permit one to search the LC-MS/MS data against a large number of potential sequences, such as all possible RNA transcripts present in a cell (Jiang et al., 2019). The development of software tailored specifically for mapping modifications identified in LC-MS/MS data against genomic and transcript data from any organism is a critical need for bottom-up modification mapping to become widely used.
Fifth, LC-MS platforms, especially those with performance characteristics required for the accurate identification of modifications within larger RNA sequences, are expensive, require trained operators, and are best suited for a dedicated facility. The development of benchtop systems with
the capabilities and functionalities to meet the needs of RNA modification mapping would broaden the availability and equitability of those who could take advantage of this technology. Furthermore, the current LC-MS approaches tend to be quite low throughput. A single LC-MS analysis, including column (re-)equilibration time, takes at least 60 minutes and often far longer (Bommisetti and Bandarian, 2022; Hagelskamp and Kellner, 2021; Jones et al., 2023). Therefore, identifying ways to generate MS/MS data more rapidly, combined with advances in data processing, will be vital for this field.
Top-down modification mapping. A less common but potentially powerful mass spectrometry method for RNA modification mapping is the so-called top-down approach. Unlike the bottom-up approach, top-down approaches use specialized mass spectrometry instrumentation to collect MS/MS data from large oligonucleotides or intact RNAs; enzymatic or chemical digestion is not required. Because the top-down approach preserves the complete sequence information, it significantly enhances the accuracy of mapping modifications (Taucher and Breuker, 2010, 2012).
The top-down approach uses a variety of gas-phase dissociation methods to generate sequence information from the sample. For example, collision-induced dissociation can provide nearly full-sequence coverage, but it requires a low precursor ion charge state, which decreases instrument performance, specifically its sensitivity and mass resolving power (Glasner et al., 2017; Taucher and Breuker, 2010).
Alternative-ion dissociation methods have been developed, many of which could enable more sensitive mapping of RNA modifications. Examples include radical transfer dissociation, activated-ion negative electron transfer dissociation, ultraviolet photodissociation and activated-electron photo-detachment dissociation (Calderisi, Glasner, and Breuker, 2020; Peters-Clarke et al., 2020; Santos et al., 2022). Each of these dissociation methods has their strengths and drawbacks, but exploration into these alternatives has demonstrated that enhanced sequence coverage during a top-down analysis is feasible.
Another advantage of top-down analyses is that they are directly compatible with native mass spectrometry, whereby both sequence and structural information can be obtained about the RNA of interest. An inherent limitation of top-down methods is that they are most effective when working with single RNA species. Their advantages may be better realized when seeking confirmation of expected modified RNAs versus discovery of unknown modifications from mixtures of RNAs.
Computational and Modeling Methods for Sequencing Technologies
As discussed in the previous section, computational methods for LC-MS/MS sequencing and modification mapping are lagging. The computational methods used for indirect sequencing are better developed than those for direct sequencing approaches. The tools available for indirect sequencing depend on quality control, alignment, and peak calling steps to localize modifications. Several of these steps have been researched extensively, with multiple tools developed for each of these steps that are shown to perform well. This section focuses on the current computational tools that are used when sequencing RNA and mapping its modifications with nanopore sequencing.
Data analysis is an integral component of the nanopore sequencing and mapping process, because the raw output is a set of changes in electrical current rather than a sequence of specific and modified bases. The major steps involved in analyzing data generated from nanopore direct RNA sequencing can be divided into basecalling, quality control, assembly and alignment, and signal data extraction (Figure 3-5).
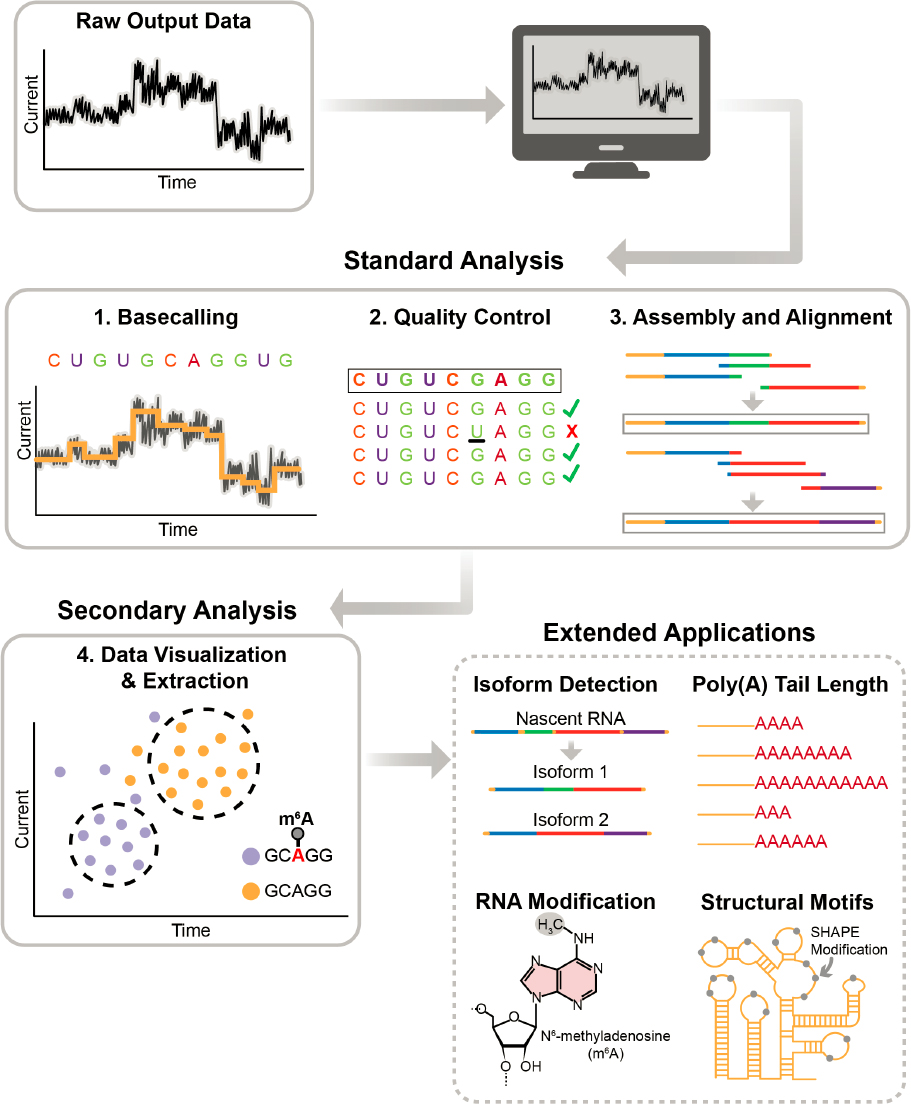
SOURCE: Secondary Analysis (bottom left) adapted from Koonchanok et al., 2021. Licensed under Creative Commons Attribution 4.0 International License.
Basecalling
Basecalling is the process of translating raw electrical signals into accurate nucleic acid base sequences. Nanopore sequencing uses a sequencing-by-translocation approach, in which the sequencer detects changes in electrical signals as different bases pass through the nanopore. Basecalling is a critical step in nanopore sequencing because noisy data from single molecules lead to high error rates (Wick, Judd, and Holt, 2019). The use of neural network–based basecallers, such as Guppy,6 Albacore,7 and Scrappie,8 has improved accuracy compared with earlier hidden Markov model approaches (Box 3-2) (Wick, Judd, and Holt, 2019). Despite continued improvements in basecalling algorithms, nanopore sequencing continues to have higher error rates than indirect sequencing techniques such as Illumina sequencing (Napieralski and Nowak, 2022; Zhang et al., 2020). Several basecalling tools have been developed, which are described in Appendix A, Table A-1.
BOX 3-2
Bioinformatics of Nanopore Sequencing
To obtain the sequence of input molecules in nanopore-based sequencers from Oxford Nanopore Technologies (ONT), an electrical signal must be translated into individual bases through a process called basecalling. MinKNOW, the software used for sequencing, offers an built-in basecaller for real-time basecalling during the sequencing run. Users can choose to turn off this function and perform basecalling on the raw files using an alternative basecaller at a later time. ONT currently offers multiple versions of the basecalling software, including Doradoa and Bonito,b which can process both DNA and RNA sequencing datasets.
Earlier versions of basecallers required that the raw current signal measurements, represented by the “squiggle” plot, be segmented into events. Each event’s duration, mean, and variance formed the input for the basecalling algorithm (Garalde et al., 2018). In the quest to develop more accurate algorithms, ONT and other research groups have been exploring machine learning–based approaches.
The initial generation of nanopore basecallers relied on hidden Markov models (HMM) to predict DNA sequences (Eddy, 2004). HMMs are probabilistic models that predict unobserved events based on the information about the previous events. However, basecallers using recurrent neural networks (RNNs) (Boža, Brejová, and Vinař, 2017) have replaced HMM-based approaches. RNNs utilize a longer range of information, and bidirectional RNN models consider bases both before and after a nucleotide of interest, leading to increased accuracy (Rang, Kloosterman, and de Ridder, 2018).
With the release of Guppy (Wick et al., 2019) and subsequent versions, which share basecalling features with previous versions of the basecallers but run on a graphics processing unit (GPU) instead of a central processing unit (CPU), basecalling speed increased more than tenfold (~1,500,000 base pairs/second versus ~120,000 base pairs/second). Guppy (V3.4.5) also reduced the basecalling error rate to between 4 and 6 percent for DNA and 7 and 12 percent for RNA (Wick et al., 2019). The latest version of the Guppy basecalling framework (as of the writing of this report) is trained to call the modified bases in DNA, 5mC and 6mA (N6-methyldeoxyadenosine), from the raw signal data.
Both HMM and RNN models rely on training signals, and basecaller performance depends on the quality of the training dataset. The significant advantage of nanopore direct RNA sequencing is the ability to access single-molecule sequencing of mRNA transcripts and investigate RNA alterations. As basecalling algorithms improve and error rates decrease, it becomes possible to revisit older raw datasets for sequencing and analyze them again using newer and more accurate algorithms.
__________________
a See https://github.com/nanoporetech/dorado (accessed November 12, 2023).
b See https://github.com/nanoporetech/bonito (accessed November 12, 2023).
___________________
6See https://timkahlke.github.io/LongRead_tutorials/BS_G.html (accessed November 12, 2023).
7See https://nanoporetech.com/about-us/news/new-basecaller-now-performs-raw-basecalling-improved-sequencing-accuracy (accessed November 12, 2023).
8See https://github.com/nanoporetech/scrappie (accessed November 12, 2023).
Quality Control
Quality control analysis of ONT sequencing data is critical to offset the error-prone basecalling that ONT instruments generate, which if left unfixed will affect downstream analysis. Some popular quality control tools for ONT long-read data include Poretools, poRe, NanoOK, ToulligQC, and PyPore. Each offers unique features and functionalities. For example, Poretools and poRe provide yield plots, histograms, and various summary statistics of MinION output (Loman and Quinlan, 2014; Watson et al., 2014). NanoOK focuses on alignment-based quality control and error profile analysis (Leggett et al., 2015). ToulligQC supports barcoding samples and produces various graphical representations for data exploration9 and PyPore improves existing tools for fast, accurate quality control (Semeraro and Magi, 2019). For a more extensive list of quality control tools, see Appendix A, Table A-2.
Sequence Assembly and Alignment
Sequencing assemblers designed for analyzing long-read sequencing data from direct sequencing platforms are crucial tools for building the end-to-end sequence of an RNA molecule from the basecalled data. A few assemblers are provided briefly as examples, while an extended list of assemblers is in Appendix A, Table A-3.
MaSuRCA is a long-read assembler that generates an error-free “super read” by joining multiple reads from the reference genome (Zimin et al., 2013). Canu is an assembler designed for noisy long-read sequences that uses a hierarchical assembly pipeline to produce accurate assemblies (Koren et al., 2017). Unicycler is a hybrid assembler that combines Illumina and ONT data for bacterial genomes (Wick et al., 2017). HINGE addresses the challenges of graph assembly with longer reads, automating the process and reducing problems with fragmented and unresolvable repeats (Kamath et al., 2017).
Alignment tools work together with assemblers to identify the correct location in the genome of a particular fragment of sequenced RNA. Appendix A, Table A-4 summarizes alignment tools used in long-read sequencing analysis, particularly those used with ONT-generated reads. Among the options are Minimap2, which is used to align long nucleotide sequences and performs well with sequences of 100 or more base pairs. GraphMap2 is a splice-aware RNA sequencing algorithm that maps long-read data to reduce repetitive alignments.10 mapAlign is a software package that includes algorithms for mapping and local alignments of long reads against a reference genome (Yang and Wang, 2021).
While long-read sequencing has advanced since direct sequencing was first introduced, raw data accessibility remains a significant challenge. Selecting the right assembler and aligner depends on the specific needs of the researcher, lab resources, and dataset characteristics. For example, Shasta is a fast assembler of nanopore reads, but it requires large computer memory (Marx, 2021; Shafin et al., 2020).
Signal Data Extraction
The raw electrical signals that are the output from a nanopore sequencer contain embedded data corresponding to the sequenced nucleotides. Thus, careful extraction and analysis of the data are essential for revealing the biological information hidden in these signals (Figure 3-5). Some of the biological information that can be gleaned is the detection of RNA isoforms, estimation of poly(A) tail length, detection of RNA modifications, and prediction of RNA secondary structures depending
___________________
9See https://github.com/GenomicParisCentre/toulligQC (accessed November 14, 2023).
10See https://github.com/lbcb-sci/graphmap2 (accessed November 14, 2023).
on the specific variant of direct RNA sequencing protocol employed. Some tools used for extraction and analysis of raw signal data are Nanopolish, Racon, Tombo, SquiggleKit, and Sequoia.
Nanopolish improves the consensus accuracy of an assembly of nanopore sequencing reads. It can detect methylation, single nucleotide variants, and insertions and deletions; align signal-level events; and estimate polyadenylated tail lengths (Loman, Quick, and Simpson, 2015). Racon is used to correct raw contigs generated by rapid-assembly methods (Vaser et al., 2017). Tombo enables the investigation, detection, and visualization of modified DNA and RNA nucleotides using statistical tests to compare signal differences (Stoiber et al., 2017). SquiggleKit can extract, process, and plot raw nanopore signal data (Ferguson and Smith, 2019). Sequoia is a visual analytics tool that allows interactive exploration of nanopore sequences, including clustering sequences based on electrical current similarities, drilling down into signals to recognize properties of interest and comparing signal features from RNA modifications (Koonchanok et al., 2021, 2023).
Computational Tools for Mapping RNA Modifications
While direct RNA sequencing offers great promise for identifying RNA modifications at single-base resolution, interpreting raw signals corresponding to modified and unmodified base-sequence contexts is challenging. Currently, more than 170 RNA modifications have been reported in the literature (Kadumuri and Janga, 2018), and generating unbiased training models encompassing these diverse modifications would imply an increase in the RNA alphabet to 174 from the mere 4 regular nucleotides currently studied. For instance, developing algorithms that can simultaneously infer four regular nucleotides and give RNA modification types from ONT data may require at least 95 possible pentamer signals to construct reference oligonucleotide signatures. (Note that ONT’s tools “see” a pentameric stretch of nucleotides in a pore.) Thus, two challenges arise: (1) capturing all of the pentamer nucleotide configurations that contain the four canonical nucleotides and the modified nucleotides observed across all of the transcriptomes of interest; and (2) generating synthetic signals corresponding to those configurations. Surmounting these two challenges will likely drive development of the next generation of nanopore RNA-modification-aware basecallers for ONT data. Table 3-3 shows a list of algorithms and software tools currently available for mapping RNA modifications using ONT-generated datasets.
Four approaches are used to predict the modifications:
- Employing basecalling errors at the modification site as a surrogate to mapping the modified nucleotides onto transcripts.
- Using RNA modification loci reported from antibody-based methods to develop modification-specific basecallers for direct RNA-sequencing data.
- Making a direct comparison of signals from wild-type and knockout cells at bonafide modification locations reported in the literature to develop machine learning models. This approach assumes that modifications are lost in cell lines when known modification writers are knocked out.
- Employing synthetically designed RNAs containing modified nucleotides to generate nanopore signals and using the resulting data for developing machine learning models that can distinguish a particular modification from its unmodified counterpart.
Most of the platforms that predict and localize modifications use machine learning models. These models depend on high-quality training datasets to perform effectively. For example, Nanocompore was developed to study RNA modifications from direct RNA sequencing data. It runs an automated pipeline for data preprocessing, including basecalling; alignment using Minimap2; and quality control using Nanopolish. Nanocompore uses an unmodified control sample to detect
TABLE 3-3 Overview of Various Tools for Identifying RNA Modifications in Direct RNA Sequencing Data
NOTE: Abbreviations are defined in the Front Matter.
transcriptome-wide m6A modification events. The developers have suggested that this framework could be used for detecting any type of RNA modification event (Leger et al., 2021).
MAJOR CHALLENGES AND SCIENTIFIC GAPS
A complete understanding of all RNA modifications—that is, where they map on each RNA molecule and their roles in living systems—will require significant technological advancement. Despite the progress that has come from adapting existing DNA and protein sequencing and analysis methods to sequence RNA and its modifications, tools and methods specific to RNA lag behind those for DNA and proteins significantly. This is, in part, because of challenges related to the intrinsic nature of RNA molecules. First, RNA molecules are relatively unstable and prone to
degradation, so they are difficult to isolate, preserve, and analyze. Second, RNA modifications are transient, and their numbers and types are specific to the microenvironment of a particular molecule, cell, or tissue, making it difficult to isolate enough copies for analysis. Third, the central dogma of molecular biology had, for a time, established RNA as a simple carrier of information from DNA (the source code) to proteins (the product). This paradigm overlooked the extraordinary complexities and functions of RNAs and their modifications, leading to the relative dearth of RNA-specific tool developments. While much has been discovered about the myriad roles of RNA and RNA modifications, this emerging field would benefit from tools and technologies dedicated to addressing the specific challenges that come with sequencing RNA and identifying, mapping, and quantifying RNA modifications.
The ultimate technological goal is to develop robust, reproducible, and accessible tools and methods for identifying all RNA modifications from one end of a single RNA molecule to the other, at a sensitivity that allows interrogation of a single cell, as well as tools and methods for determining modification stoichiometry and crosstalk between multiple modifications within a single RNA. Several limitations in the currently available technologies for global modification measurement, indirect sequencing, direct sequencing, computational tools, and databases prevent accomplishing this ultimate goal. As discussed in the preceding sections, limitations for each of the available technologies for interrogating RNA modifications include the following:
- The use of LC-MS/MS for the global characterization and quantification of modified nucleosides (or nucleotides) remains ripe for improvement. For one, the approach cannot yet quantify all modifications in complex mixtures with both high accuracy and high precision. Beyond those described earlier in this chapter, new, mass spectrometry–friendly ways to process RNA samples with minimal sample handling would reduce sample loss or degradation, and in turn, would reduce the amount of RNA needed for the analysis. Unique derivatization reagents applicable to all modified nucleosides that take advantage of mass spectrometry ionization and detection methods could enhance sensitivity, which would also reduce initial sample requirements.
- Next-generation indirect sequencing generates a large volume of data at low cost but cannot yet do so at the single-molecule or single-cell level. It also does not allow simultaneous interrogation of modifications. Third-generation indirect sequencing is amenable to single-molecule and single-cell DNA sequencing, but applications for RNA have been explored only cursorily.
- Direct sequencing holds the most promise for RNA, but the options are still in early developmental stages and are therefore expensive, require substantial amounts of starting material, and are prone to error.
Addressing gaps in measurement technologies would have maximal impact if accompanied by addressing gaps in the associated computational tools that allow analysis, interpretation, and use of the data generated. Computational methods for improving the basecallers used by many direct sequencing methods are critical for improving the ability of these sequencers to detect modified RNA nucleotides accurately. Current basecallers can recognize the signals generated by only a handful of the ~170 RNA modifications. Furthermore, it can be difficult or impossible to distinguish between some modifications, such as N1-methyladenosine (m1A) and m6A, in certain contexts.
To date, progress has been very limited in developing algorithms that can detect simultaneously the co-occurrence, interaction, and functional consequences of one or more RNA modifications at single-molecule resolution. The ability to detect or infer 10–15 different modifications on a single RNA molecule simultaneously would be a significant step forward. Using artificial intelligence and machine learning methods to develop and train an ensemble basecaller could help in achieving this
advance (see Emerging Computational Tools). In addition, the capability to synthesize long RNA oligonucleotides with these modifications in a variety of combinations and sequence contexts (see Chapter 4) will be needed for training these methods.
Tools that can visualize or simulate the impact of RNA modifications on the two- and three-dimensional structure of RNA molecules will move the field closer to understanding the functional role of every modified RNA isoform. Determining the impact of RNA modifications on the higher-order structure of an RNA molecule could be aided by better tools for RNA structure visualization. Embedding such tools in genome and RNA-specific browsers would enable researchers to look at RNA molecules as they may occur in the cell, rather than as a linear array of ribonucleotides. These tools could also make it possible to view or simulate structures with and without modifications present, thus giving complementary insights into the relationship between specific modifications and RNA structure; current tools, designed mostly in the style of linear DNA representations, are unlikely to provide these insights.
Currently, when interrogating an RNA sequence, most reference data are derived from genomes (i.e., DNA-based reference data). It will be important to develop RNA-based reference data to better enable researchers to query databases about RNA structure–function relationships, while sharing a common coordinate system with the DNA-based reference data for convenient cross-referencing.
EMERGING TOOLS AND TECHNOLOGIES
One of the major lessons learned from other large-scale biotechnological efforts, most notably the Human Genome Project, is that focused and concerted organization and funding directed toward a common goal accelerates technological innovation. While it is difficult to predict the emergence and trajectory of breakthrough technologies, this section explores some intriguing concepts that are shaping the future of research and development in the field of RNA modifications.
To achieve the ultimate technological goal of identifying all RNA modifications in an RNA molecule and determining their stoichiometry and crosstalk, major advancements are needed in several general categories.
First, the technologies must be sensitive enough to analyze a single molecule and specific enough to deduce the sequence and composition of each ribonucleotide within that sequence. Achieving high sensitivity and high specificity will likely come from improved sensors, signal processing, and analysis tools, as well as advancements in RNA sample preparation methodologies, including purification/concentration methods and possibly RNA amplification methods that preserve modification information. For instance, RNA-dependent RNA polymerases—which have the ability to catalyze the synthesis of an RNA strand complementary to a given RNA template and are found across a range of organisms, including viral and plant genomes—could contribute to improved methods for amplifying RNA (Dalmay et al., 2000; Gerlach et al., 2015; Shu and Gong, 2016; Wu and Gong, 2018; Yin et al., 2020). Hence, RNA amplification methods, especially those that can preserve the modifications or modification-related information, could significantly circumvent the challenges associated with the need for large input material in direct RNA-sequencing protocols.
Second, the technologies must have the flexibility to sequence the diversity of RNA inputs, including short and long RNA sequences; mitigate the effects of secondary and tertiary structures; and accept sample inputs from a variety of biological sources.
Third, the technologies must be robust and widely accessible. Current workflows are cumbersome and require highly skilled users or teams to perform them. Future innovations must offer turn-key solutions to empower a broader research community to work with RNA.
Fourth, there is a need for advancements in computational tools and databases that are specific for the study of RNA and its modifications.
And lastly, as discussed in the next chapter, these technologies must be complemented with advancements in the preparation, stability, and accessibility of a wide range of RNA references and standards that represent the diversity of RNA modifications and that satisfy the needs of RNA sequencing technologies.
While there is some possibility that these goals will be achieved by a singular sequencing technology, it seems likely, particularly in the short term, that a combination of complementary, crosscutting, and hybrid approaches will be required. The following sections highlight some promising emerging technologies that (1) capitalize on existing DNA and protein sequencing technologies, (2) adapt existing structural biology technologies, and (3) leverage technologies that are not traditionally associated with biological analysis.
Emerging Indirect Sequencing Technologies
The development of reagents and procedures that derivatize RNA in a modification-specific manner will enable indirect sequencing methods to be used for end-to-end detection and sequencing of modified RNAs. For example, several microbial enzymes have been found to alter modified nucleotides in ways that mark these nucleotides during the reverse transcription process that precedes sequencing. Escherichia coli AlkB demethylase and its engineered derivatives can remove methylations that interfere with reverse transcription from tRNA and mRNA molecules prior to sequencing library construction (DM-tRNA-seq, ARM-seq) (Cozen et al., 2015; Zheng et al., 2015). E. coli endoribonuclease MazF can cleave an unmethylated 5′-ACA-3′motif, but not the 5′-m6ACA-3′motif, which when followed by NGS enables those m6A nucleotides to be identified (MAZTER-seq [Garcia-Campos et al., 2019], m6A-REF-seq [Zhang et al., 2019]). The rRNA modification enzyme Mjdm1 can selectively label m6A-modified nucleotides in the transcriptome (m6A-SAC-seq) (Hu et al., 2022). The evolved enzyme TadA can convert unmodified A into inosine, which is read as G during sequencing, while m6A nucleotides read as A when sequenced (eTAM-seq) (Xiao et al., 2023).
Another source of protein reagents useful for RNA modification sequences is RNA-binding protein domains, which recognize specific modifications, such as the human YTH domain used in the DART-seq (deamination adjacent to RNA medication targets) method of m6A sequencing (Meyer, 2019). Additionally, engineered antibodies that recognize specific modifications have been a useful tool for modification mapping; examples include antibodies against m6A, N1-methyladenosine (m1A), and N7-methylguanosine (m7G) (Table 3-1) (Helm and Motorin, 2017; Zhang, Lu, and Li, 2022). These examples demonstrate the utility of protein agents for RNA modification sequencing and highlight the need for continued efforts to identify new and novel enzymes and modification readers from microbial and eukaryotic sources.
CRISPR-based systems have also been used to target specific RNAs by expressing RNA-specific guide RNAs together with RNA-binding Cas proteins. For instance, fusing catalytically inactive Cas13 (dCas13) with human ADAR2 has been used to direct A-to-I editing in RNAs of interest (Cox et al., 2017). This strategy enables selective alteration of protein composition and has potential utility as a therapeutic agent for correcting disease-causing variants. Similarly, tethering dCas13 to the METTL3/METTL4 methyltransferase complex proteins can be used to achieve site-specific m6A modification of target RNAs (Wilson et al., 2020). Targeted m6A removal has also been demonstrated by fusing catalytically dead Cas proteins to RNA demethylase enzymes (Li et al., 2020; Liu et al., 2019b). These examples highlight the utility of CRISPR-based approaches for enabling selective chemical manipulation of RNAs of interest. Such systems have tremendous potential to be used both as basic biochemical research tools and as novel therapeutics, but require that proper controls be developed to evaluate them and their limitations before they can be used for therapeutic benefit. Therefore, continued engineering of improved CRISPR systems that have
enhanced specificity and sensitivity for different types of modifications will be critical for expanding the capabilities of these systems.
The advantage of marking modified nucleotides within the copied DNA following reverse transcription is that currently available long-read (e.g., PacBio) and single-cell sequencing techniques (e.g., 10x Genomics) can be used to map specific modifications along the entire length of an RNA and in single cells, respectively. Unfortunately, it may prove impossible to develop specific reagents for every modification, considering that some modifications are similar in structure and reactivity and that others may not be amenable to such approaches due to their chemistry. Raising antibodies against specific modifications will also contribute to the expansion of indirect sequencing approaches. These will continue to require rigorous quality control during production and use to avoid misinterpretation of false-positive signals that frequently result from cross-reactivity with other antigens, such as the antibody raised against m1A, which shows affinity for the m7G cap structure (Grozhik et al., 2019).
Emerging Direct Sequencing Technologies
Sequencing by Mass Spectrometry
Although mass spectrometry is considered the gold standard for the global analysis of RNA modifications at the nucleoside level, utilizing mass spectrometry for direct sequencing of RNA and its modifications—especially RNAs longer than 150 mers—is challenging. Developers and vendors of mass spectrometry platforms recognize the importance of this technology for the characterization of modified RNAs. While recent commercial developments have emphasized LC-MS/MS methods for confirming and characterizing RNA therapeutics and other RNA-based biological tools such as guide RNAs (Macias et al., 2023), these advances can be expanded to more general applications in RNA sequencing of biological materials. For example, end-to-end sequencing of tRNAs at levels approaching single molecules will require significant gains in sensitivity and reductions in sample losses. It would be valuable to focus research and development on instrument platforms developed specifically for detection of negatively charged species, as mass spectrometry of oligonucleotides is presently about 100 to 1,000 times less sensitive than is mass spectrometry for peptides and proteins. One way to drive this kind of innovation would be to encourage and incentivize partnerships between vendors and academic labs that are pushing the boundaries of fundamental research and discovery in mass spectrometry.
Opportunities also exist for improving the front-end separation methods required when mapping RNA modifications by mass spectrometry. HPLC, the current method of choice, is limited primarily to ion-pairing reversed-phase approaches (see the previous section, Mass Spectrometry-based Methods). Optimizing additional separation approaches for use with RNA samples, such as ion mobility, size exclusion chromatography, and capillary electrophoresis, to be used alone or in combination with existing separation methods, could improve both sequencing read lengths and sequence coverage depth by mass spectrometry. Integrated methods that enable on-line purification of oligonucleotides prior to separation and mass spectrometry analysis are examples of technology developments that can close existing gaps for this field.
Building upon examples that utilize mass spectrometry and other sequencing technologies, there are opportunities to create more sensitive analytical workflows that combine the strengths of indirect sequencing methods and mass spectrometry for higher-quality RNA modification maps (de Crécy-Lagard et al., 2020; Kimura, Dedon, and Waldor et al., 2020; Puri et al., 2014). These hybrid approaches will likely be most powerful for analyzing RNAs that possess a significant diversity in modification types (e.g., tRNAs and some therapeutic RNAs). Success here will most likely require supporting one or more core RNA sequencing centers where the combined expertise can
coexist and services can be utilized by the broader research community. For instance, such centers would benefit from having sequencing, mass spectrometry, and computing capabilities in house, in addition to the expertise needed to analyze and interpret the data and apply various computational and bioinformatics techniques.
Sequencing by Nanopore
As demonstrated by the success of ONT’s nanopore sequencing, biological pores hold great promise for detecting RNA modifications with single-molecule resolution. A large number of biological and solid-state nanopores have been discovered and shown to have the capability to sense a wide array of biological molecules, including DNA, RNA, and proteins (Brown and Clarke, 2016; Mayer, Cao, and Dal Peraro, 2022; Xue et al., 2020). While the majority of the work toward detecting RNA modifications by nanopore sequencing has been completed using biological nanopores, solid-state nanopores have emerged with the promise of being robust and tunable alternatives to their biological counterparts (Dekker, 2007; Henley, Carson, and Wanunu, 2016). However, direct detection of RNA modifications using either of these nanopore technologies remains a significant challenge (Abebe, Verstraten, and Depledge, 2022; NASEM, 2023).
Modified biological or engineered solid-state pore structures have the potential to expand the dynamic range of nanopore signal intensity or dwell time for improving noise and reducing basecalling errors. For example, a novel biological pore structure was successfully engineered to detect seven modified RNA mononucleosides in addition to the canonical RNA mononucleosides (Wang et al., 2022). While this approach represents progress toward the ultimate goal of quantitative end-to-end sequencing of all RNA modifications, it remains limited to a relatively small subset of RNA modifications and samples with high copies of RNA inputs (Punthambaker, 2022). Further development of biological and solid-state pores with high sensitivity and low noise, as well as continued training and optimization of algorithms with larger datasets, is needed to continue to advance nanopore technology.
Direct detection of RNA modifications by nanopore sequencing can also be used in combination with high-throughput chemical probing approaches to characterize RNA structure (Stephenson et al., 2022). When RNA molecules are exposed to reagents that chemically modify the RNA in a structure-dependent fashion, detection of these modifications gives information about RNA structure (Spitale and Incarnato, 2023). While much work has been invested in using indirect sequencing methods to reverse transcribe modified RNAs and then analyze the resulting cDNAs (Strobel, Yu, and Lucks, 2018), recent efforts have focused on using nanopore technology to directly detect chemical probe adducts (Bohn et al., 2023) and both endogenous RNA and chemical probing modifications (Stephenson et al., 2022). For example, chemically assisted nanopore sequencing using the bisulfite reaction has been shown to be useful in high-confidence assignment of Ψ modification because of the unique signature of the Ψ–bisulfite adduct (Fleming et al. 2023). Further, cross-disciplinary collaboration between the nanopore sequencing community and the RNA structure-chemical probing field could yield ways to create defined standards to study RNA modification and RNA structures. Progress in this area could also be used to study the relationship between RNA modifications and RNA structures (Liu et al., 2015; Spitale et al., 2015).
Taken together, these emerging nanopore technologies demonstrate early, but promising, steps toward meeting the goals set forth in this report. Further investment and research will be needed to develop these technologies into robust and scalable approaches.
Sequencing by Nontraditional Technologies
One of the greatest challenges with sequencing RNA modifications is the large number of modifications that have been reported, more than 170 currently. While approaches such as chemical/enzymatic derivatization, fluorescence labeling, antibody detection, and bioelectrical sensing have been developed to detect a few specific RNA modifications each, it is unlikely that they will be able to detect all these modifications alone or in combination, even with further improvements to these methods. Therefore, new technologies that expand the ability to detect all modifications will be needed. One promising candidate is surface-enhanced Raman spectroscopy (SERS) (Brueck, 2023). Early work has demonstrated that SERS is capable of detecting DNA, RNA, and modified nucleotides at single-molecule resolution. If SERS can be combined with a system for channeling a single RNA molecule through a nanopore, it has the potential to unlock the end-to-end sequencing of many, if not all, modifications within the same RNA.
Furthermore, advancements in complementary metal oxide semiconductor sensors (CMOS) offer opportunities to develop DNA and RNA sequencing approaches that are both lower in cost and more scalable (Mola, 2023) than currently available technologies. CMOS sensors are cheap to manufacture and energy efficient, and can be programmed to sense a broad array of biomolecules, including DNA, RNA, and proteins. While these technologies hold promise for sequencing of RNA and its modifications, further research and development is needed to demonstrate their applicability.
Emerging Structural Biology Technologies
Structural biology technologies—including cryo-electron microscopy, nuclear magnetic resonance, and X-ray crystallography—provide atom-level resolution; in certain cases, they have been used to identify and map modifications within short stretches of RNA (Ruiz-Arroyo et al., 2023; Yoluç et al., 2021) and even full-length RNAs (Bhatt et al., 2021). Although they have been used most often to study proteins, structural biology techniques have a venerable history of elucidating key structural and functional features of nucleic acids, including their modifications (Minchin and Lodge, 2019; Yoluç et al., 2021). X-ray crystallography played a critical role in illuminating the structure and mechanisms of nucleic acids (Egli and Pallan, 2010; Neidle and Berman, 1983)—per-haps most notably, the double helix structure of DNA in 1953 and the first structures of the ribosome in the early 2000s (Ramakrishnan, 2002). Nuclear magnetic resonance has revealed key structural and dynamic features of nucleic acids in aqueous solution, including those of tRNAs (Kasimova, Lindahl, and Delemotte, 2018).
With growing interest in RNA modifications, researchers have been applying these three structural biology techniques to better understand RNA modifications and their role in living systems. Despite difficulties in crystallizing pure and homogenous RNA, X-ray crystallography remains a technology of great interest for understanding the structure and function of RNA modifications and their interactions with proteins (Jackson, Smathers, and Robart, 2023; Ruiz-Arroyo et al., 2023). Although it is currently most effective with short RNAs, recent advances make nuclear magnetic resonance suitable for probing the temporal dynamics of RNA modifications in solution, such as the chronology of modification addition during RNA maturation (Barraud et al., 2019; Gato et al., 2021). Continued advancements in cryo-electron microscopy have improved the technique’s resolution, enabling visualization of RNA modifications within the structural context of the ribosome (Bhatt et al., 2021; Cottilli et al., 2022; Pellegrino et al., 2023) and in a tRNA–protein complex (Ruiz-Arroyo et al., 2023).
Cryo-electron microscopy requires much less material than crystallography or nuclear magnetic resonance; sample input requirements remain a constraint for the latter two approaches. Furthermore, all structural methods are currently relatively costly and time consuming, so their utility
for identifying and mapping new modifications at scale remains limited. While the primary value of structural biology technologies may be in the functional characterization and confirmation of known RNA modifications, there is promise that these methods can one day be used to determine the temporal and spatial dynamics of RNA modifications, as well as the position, function, and interactions of modifications within RNA structures.
Emerging Computational Methods
Mapping and sequencing RNA modifications requires further advances in computational tools for a number of purposes. Direct sequencing methods, such as those using nanopores, require tools that accurately basecall the nucleotides as they travel through the nanopore. Basecalling models that can accurately identify signals from many modified RNA nucleotides do not yet exist. However, some models can detect individual modifications, such as m6A, for which enough data are available to train the models (Cruciani et al., 2023). Unfortunately, training data for other modifications are limited, impeding the development of basecalling models for these RNA-modified bases. ONT has been working for some time on algorithms that accurately detect modified nucleotides, focusing on detecting methylated bases, predominantly those in DNA. ONT’s tool Remora11 is an epibasecaller that works alongside a canonical basecalling algorithm, such as Bonito.12 During a workshop hosted by the committee, it was noted that Remora is being trained on RNA training sets with a near-term goal of reading modified RNA bases at an accuracy of 95 percent (NASEM, 2023).
Machine learning and deep learning approaches are also showing promise for enabling de novo sequencing of RNA modifications. In these approaches, basecalling algorithms are trained on large datasets to differentiate error from modification signal and to make predictions; some are further optimized by implementing neural networks (Zhao et al., 2022). Deeplearning Explore Nanopore m6A (DENA) and nanopore sequencing–based isoform dynamics are two examples of methods that use deep learning models and neural networks to predict and quantify RNA modifications (Maier et al., 2020; Qin et al., 2022). It is anticipated that developing physical modified-RNA standards, generating sufficient experimental datasets for all RNA modifications, and improving the nanopore sequencing platform will further advance the range and accuracy of these methods.
In order to use mass spectrometry to detect RNA modifications during sequencing, several challenges have to be overcome to analyze and correctly align the oligonucleotide fragments that go into the instrument. The software has to be capable of analyzing complex samples containing multiple types of modifications, statistically validating the accuracy of base assignments and fragment alignments, and integrating other analytical tools. One tool that begins to address these challenges is NucleicAcidSearchEngine, a database-matching tool that identifies RNA oligonucleotide MS/MS spectra (Wein et al., 2020); it is integrated into OpenMS,13 the open-source framework for mass spectrometry.
RNA modifications impact the structure of their RNA parent molecule, which can affect interactions between the RNA molecule and RNA-binding proteins, especially if modifications are in the protein-binding region (Lewis, Pan, and Kalsotra, 2017). Thus, understanding these interactions at the structural level can help clarify the roles of specific RNA modifications and combinations of modifications in structural interactions and biological functions. Computational approaches can enable the study of RNA–protein interactions. For example, a tool has been developed that uses short molecular dynamics simulations of an RNA–protein complex of interest, along with energy calculations, to search for RNA modifications that exhibit energetically favorable interactions with
___________________
11See https://github.com/nanoporetech/remora (accessed November 12, 2023).
12See https://github.com/nanoporetech/bonito (accessed November 12, 2023).
13See https://openms.de/_ (accessed November 6, 2023).
the binding protein. That subset of modifications is then included combinatorially in the RNA, and longer molecular simulations are performed. The result is a set of RNA modifications predicted to enhance the interactions between a specific RNA molecule and its binding protein (Orts and Gossert, 2018). Hence, there is a significant need for computational tools that can account for the presence of various RNA modifications and provide an assessment of their interaction alternation with proteins, DNA, and metabolites, as well as other cellular entities.
Several companies are using artificial intelligence (AI) to evaluate RNA structures and the impact of modifications on RNA structure, using that information to design small-molecule therapeutics and to identify new drug targets. For example, ReviR Therapeutics’14 discovery process is driven by AI algorithms that analyze, model, and predict the chemical space inhabited by modified RNA and its interactions with small molecules and macromolecules in cells. ReviR uses AI to analyze large amounts of sequencing and RNA structure data to identify potential binding sites for small molecules. These potential interactions can be probed computationally, and promising candidates can be evaluated in cell-culture and animal models (McMahon, 2023). Exemplifying another approach, Atomic AI has developed a deep neural network for predicting the three-dimensional structures of RNA molecules (Townshend et al., 2021). Atomic AI’s approach predicts RNA structures with high accuracy despite being trained on a small set of known RNA structures. The company is integrating its computational results with large-scale experimental methods to design small-molecule drugs and RNA-based therapeutics. Future advances aided by AI and machine learning may include improving global prediction of RNA modifications, functional information, interactions, and integration with other biological data (Acera Mateos et al., 2023).
Although numerous advancements in computational methods for studying RNA are needed, these examples highlight the progress and innovation coming from both academic and commercial laboratories to provide the necessary computational tools.
CONCLUSIONS
Conclusion 3–1:Currently available technologies and tools for global measurements of modifications and indirect and direct sequencing of RNA cannot identify all RNA modifications, determine their stoichiometry, or elucidate crosstalk between multiple modification sites in a single RNA molecule.
Conclusion 3–2:To advance understanding of the roles of RNA and RNA modifications in living systems, it is imperative to develop technologies that can overcome the limitations described in Conclusion 3-1. These technologies must also provide reproducible results and be robust, affordable, readily available, and operable with a reasonable amount of training. Ideally, these technologies would enable sequencing of each RNA molecule in a sample from end to end; in a single experiment; and eventually, at the single-cell level.
Conclusion 3–3:While it is difficult to predict the trajectory of technological advancement, large-scale scientific efforts such as the Human Genome Project have shown that focused and concerted organization and funding directed toward a set of well-defined goals will accelerate technological innovation in that field. The committee anticipates that a similar effort focused on the goal of sequencing and mapping RNA modifications will speed up technological innovations in this field.
___________________
14See https://revirtx.com (accessed November 6, 2023).
ROADMAP FOR ADVANCING TOOLS AND TECHNOLOGIES
The following general timeline and order of operations are based on the committee’s priorities. The order of the items under each time frame is more important than the number of suggested years. Figure 3-6 compiles the roadmap’s actions and milestones for advancing tools and technologies in pursuit of mapping any epitranscriptome.
Within 5 years:
- Standardize sample isolation, preparation, and handling methods for global analyses.
- Establish RNA core centers that offer a range of expertise, techniques, and methods spanning the sequencing approaches (e.g., global modification mapping, direct and indirect sequencing), as well as the computational and bioinformatics expertise needed to analyze and interpret the data.
- Refine and optimize existing approaches.
- Improve the sensitivity and specificity of instrumentation used for cataloging and quantifying all RNA modifications in a single sample. Develop new enzymes and chemicals to facilitate improved sensitivity and specificity of modification detection.
- Refine experimental and corresponding computational approaches for indirect and direct sequencing, such as basecalling algorithms, that can simultaneously determine the presence and quantity of multiple RNA modifications with high accuracy and precision.
- Begin research to extend these capabilities to the single-cell level, which will enable dynamic understanding of specific modifications without averaging over different cell types or cellular states. Improving sensitivity of methods and compatibility of workflows will be important for accomplishing this.
- Compile and centralize existing information about available technology, tools, and methods and associated computational toolkits.
- Compile resources for informing users about the strengths, weaknesses, and potential biases of different methods.
- Centralize the databases and make software tools easily accessible for both experienced and first-time epitranscriptomics researchers and clinicians.
- Explore novel approaches.
- Promote the development of new ideas and new technologies for sequencing RNA modifications by providing forums and opportunities for innovative, cross-disciplinary thinking.
- Expand opportunities for collaborations and partnerships among academic, industrial, and government sectors.
- Encourage innovation in research by investing in novel approaches for epitranscriptomics.
- Map modifications for abundant RNAs, such as tRNAs and rRNAs, for multicellular eukaryotic organisms in single, well-defined states, including stoichiometry.
- Determine epitranscriptomes for viral pathogens, such as SARS-CoV-2, HIV, and influenza.
Within 10 years:
- Establish high-throughput, low-cost, highly sensitive clinical assays for multiple RNA modifications relevant to human health and disease.
- Facilitate broad access to epitranscriptome sequencing by driving down costs and improving usability.
- Accurately measure changes in modification stoichiometry across multiple defined cellular conditions for RNAs with high modification diversity (e.g., tRNAs).
- Understand modification crosstalk via modification measurements at the single-molecule level for RNAs with high modification diversity (e.g., tRNAs).
- Characterize epitranscriptomes of cultured cell lines and tissues in single, well-defined states.
- Complete the first epitranscriptome map of one or more simple multicellular eukaryotic organisms in a single, well-defined state.
Within 15-years:
- Enable end-to-end sequencing and mapping of all RNAs and their modifications.
- Apply tools and technologies for answering important scientific questions and providing insights into human health and disease.
- Leverage knowledge of RNA modifications for applications beyond health and medicine in other key areas, such as agriculture and synthetic biology.
- Continue to move epitranscriptomics toward single-cell and single-molecule applications.
- Complete epitranscriptomes of multiple, complex multicellular eukaryotic organisms under multiple defined cellular conditions.
- Profile RNA modifications in human diseases.
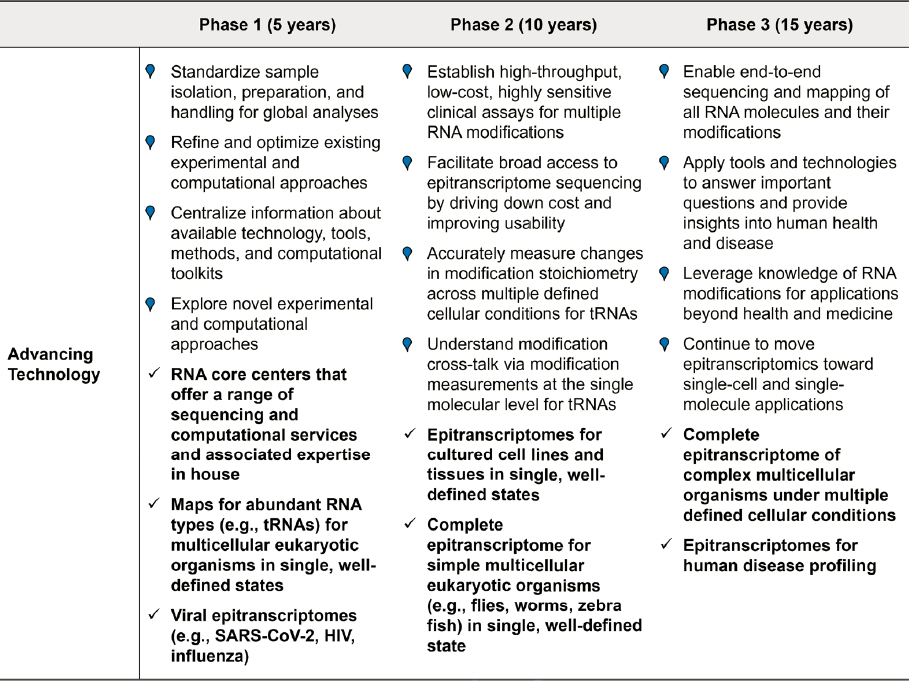
NOTES: Colored points represent actions needed for progress; check marks indicate milestones along the path for achieving the 15-year vision of mapping epitranscriptomes. tRNA = transfer RNA.
REFERENCES
Abebe, J. S., R. Verstraten, and D. P. Depledge. 2022. “Nanopore-based detection of viral RNA modifications.” mBio 13 (3): E0370221. https://doi.org/10.1128/mbio.03702-21.
Acera Mateos, P., Y. Zhou, K. Zarnack, and E. Eyras. 2023. “Concepts and methods for transcriptome-wide prediction of chemical messenger RNA modifications with machine learning.” Briefings in Bioinformatics 24 (3). https://doi.org/10.1093/bib/bbad163.
Addepalli, B., N. P. Lesner, and P. A. Limbach. 2015. “Detection of RNA nucleoside modifications with the uridine-specific ribonuclease MC1 from Momordica charantia.” RNA 21 (10): 1746–1756. https://doi.org/10.1261/rna.052472.115.
Addepalli, B., S. Venus, P. Thakur, and P. A. Limbach. 2017. “Novel ribonuclease activity of cusativin from Cucumis sativus for mapping nucleoside modifications in RNA.” Analytical and Bioanalytic Chemistry 409 (24): 5645–5654. https://doi.org/10.1007/s00216-017-0500-x.
Anreiter, I., Mir, Q., Simpson, J. T., Janga, S. C., and Soller, M. 2021. “New twists in detecting mRNA modification dynamics.” Trends in Biotechnology, 39(1), 72–89. https://doi.org/10.1016/j.tibtech.2020.06.002
Apffel, A., J. A. Chakel, S. Fischer, K. Lichtenwalter, and W. S. Hancock. 1997. “Analysis of oligonucleotides by HPLC-electrospray ionization mass spectrometry.” Analytical Chemistry 69 (7): 1320–1325. https://doi.org/10.1021/ac960916h.
Arango, D., D. Sturgill, N. Alhusaini, A. A. Dillman, T. J. Sweet, G. Hanson, M. Hosogane, W. R. Sinclair, K. K. Nanan, M. D. Mandler, S. D. Fox, T. T. Zengeya, T. Andresson, J. L. Meier, J. Coller, and S. Oberdoerffer. 2018. “Acetylation of cytidine in mRNA promotes translation efficiency.” Cell 175 (7): 1872–1886. E24. https://doi.org/10.1016/j.cell.2018.10.030.
Ayub, M., and H. Bayley. 2016. “Engineered transmembrane pores.” Current Opinion in Chemical Biology 34: 117–126. https://doi.org/10.1016/j.cbpa.2016.08.005.
Barraud, P., A. Gato, M. Heiss, M. Catala, S. Kellner, and C. Tisné. 2019. “Time-resolved NMR monitoring of tRNA maturation.” Nature Communications 10 (1): 3373. https://doi.org/10.1038/s41467-019-11356-w.
Bayley, H. 2015. “Nanopore sequencing: From imagination to reality.” Clinical Chemistry 61 (1): 25–31. https://doi.org/10.1373/clinchem.2014.223016.
Begik, O., M. C. Lucas, L. P. Pryszcz, J. M. Ramirez, R. Medina, I. Milenkovic, S. Cruciani, H. Liu, H. G. S. Vieira, A. Sas-Chen, J. S. Mattick, S. Schwartz, and E. M. Novoa. 2021. “Quantitative profiling of pseudouridylation dynamics in native RNAs with nanopore sequencing.” Nature Biotechnology 39 (10): 1278–1291. https://doi.org/10.1038/s41587-021-00915-6.
Bhatt, P. R., A. Scaiola, G. Loughran, M. Leibundgut, A. Kratzel, R. Meurs, R. Dreos, K. M. O’Connor, A. McMillan, J. W. Bode, V. Thiel, D. Gatfield, J. F. Atkins, and N. Ban. 2021. “Structural basis of ribosomal frameshifting during translation of the SARS-CoV-2 RNA genome.” Science 372 (6548): 1306–1313. https://doi.org/10.1126/science.abf3546.
Bohn, P., A. S. Gribling-Burrer, U. B. Ambi, and R. P. Smyth. 2023. “Nano-DMS-MaP allows isoform-specific RNA structure determination.” Nature Methods 20 (6): 849–859. https://doi.org/10.1038/s41592-023-01862-7.
Bommisetti, P., and V. Bandarian. 2022. “Site-specific profiling of 4-thiouridine across transfer RNA genes in Escherichia coli.” ACS Omega 7 (5): 4011–4025. https://doi.org/10.1021/acsomega.1c05071.
Borek, C., V. F. Reichle, and S. Kellner. 2020. “Synthesis and metabolic fate of 4-methylthiouridine in bacterial tRNA.” Chembiochem 21 (19): 2768–2771. https://doi.org/10.1002/cbic.202000272.
Boža, V., B. Brejová, and T. Vinař. 2017. “DeepNano: Deep recurrent neural networks for base calling in MinION nanopore reads.” PLOS One 12 (6): e0178751. https://doi.org/10.1371/journal.pone.0178751.
Brown, C. G., and J. Clarke. 2016. “Nanopore development at Oxford Nanopore.” Nature Biotechnology 34 (8): 810–811. https://doi.org/10.1038/nbt.3622.
Brueck, S. 2023. “Long-read, label-free sequencing.” Armonica Technologies, Inc. PowerPoint presentation, Information Gathering Session on Sequencing Start-Ups Focused on RNA Modifications. https://www.youtube.com/watch?v=5FSMtqUyOFM.
Cai, W. M., Y. H. Chionh, F. Hia, C. Gu, S. Kellner, M. E. McBee, C. S. Ng, Y. L. Pang, E. G. Prestwich, K. S. Lim, I. R. Babu, T. J. Begley, and P. C. Dedon. 2015. “A platform for discovery and quantification of modified ribonucleosides in RNA: Application to stress-induced reprogramming of tRNA modifications.” Methods in Enzymology 560: 29–71. https://doi.org/10.1016/bs.mie.2015.03.004.
Calderisi, G., H. Glasner, and K. Breuker. 2020. “Radical transfer dissociation for de novo characterization of modified ribonucleic acids by mass spectrometry.” Angewandte Chemie International Edition 59 (11): 4309–4313. https://doi.org/10.1002/anie.201914275.
Carlile, T. M., M. F. Rojas-Duran, B. Zinshteyn, H. Shin, K. M. Bartoli, and W. V. Gilbert. 2014. “Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells.” Nature 515 (7525): 143–146. https://doi.org/10.1038/nature13802.
Chen, L., L. S. Zhang, C. Ye, H. Zhou, B. Liu, B. Gao, Z. Deng, C. Zhao, C. He, and B. C. Dickinson. 2023. “Nm-Mutseq: A base-resolution quantitative method for mapping transcriptome-wide 2'-O-methylation.” Cell Research 33 (9): 727–730. https://doi.org/10.1038/s41422-023-00836-w.
Cheng, M. Y., X. J. You, J. H. Ding, Y. Dai, M. Y. Chen, B. F. Yuan, and Y. Q. Feng. 2021. “Novel dual methylation of cytidines in the RNA of mammals.” Chemical Science 12 (23): 8149–8156. https://doi.org/10.1039/d1sc01972d.
Clark, K. D., S. S. Rubakhin, and J. V. Sweedler. 2021. “Single-neuron RNA modification analysis by mass spectrometry: Characterizing RNA modification patterns and dynamics with single-cell resolution.” Analytical Chemistry 93 (43): 14537–14544. https://doi.org/10.1021/acs.analchem.1c03507.
Clark, W. C., M. E. Evans, D. Dominissini, G. Zheng, and T. Pan. 2016. “tRNA base methylation identification and quantification via high-throughput sequencing.” RNA 22 (11): 1771–1784. https://doi.org/10.1261/rna.056531.116.
Cottilli, P., Y. Itoh, Y. Nobe, A. S. Petrov, P. Lisón, M. Taoka, and A. Amunts. 2022. “Cryo-EM structure and rRNA modification sites of a plant ribosome.” Plant Communications 3 (5): 100342. https://doi.org/10.1016/j.xplc.2022.100342.
Cox, D. B. T., J. S. Gootenberg, O. O. Abudayyeh, B. Franklin, M. J. Kellner, J. Joung, and F. Zhang. 2017. “RNA editing with CRISPR-Cas13.” Science 358 (6366): 1019–1027. https://doi.org/10.1126/science.aaq0180.
Cozen, A. E., E. Quartley, A. D. Holmes, E. Hrabeta-Robinson, E. M. Phizicky, and T. M. Lowe. 2015. “ARM-seq: AlkB-facilitated RNA methylation sequencing reveals a complex landscape of modified tRNA fragments.” Nature Methods 12 (9): 879–884. https://doi.org/10.1038/nmeth.3508.
Crain, P. F., J. D. Alfonzo, J. Rozenski, S. T. Kapushoc, J. A. McCloskey, and L. Simpson. 2002. “Modification of the universally unmodified uridine-33 in a mitochondria-imported edited tRNA and the role of the anticodon arm structure on editing efficiency.” RNA 8 (6): 752–761. https://doi.org/10.1017/s1355838202022045.
Cruciani, S., A. D. Tejedor, L. Pryszcz, R. Medina, L. Llovera, and E. M. Novoa. 2023. “De novo basecalling of m6A modifications at single molecule and single nucleotide resolution.” bioRxiv. https://doi.org/10.1101/2023.11.13.566801.
Cui, J., Q. Liu, E. Sendinc, Y. Shi, and R. I. Gregory. 2021. “Nucleotide resolution profiling of m3C RNA modification by HAC-seq.” Nucleic Acids Research 49 (5): E27. https://doi.org/10.1093/nar/gkaa1186.
D’Ascenzo, L., A. M. Popova, S. Abernathy, K. Sheng, P. A. Limbach, and J. R. Williamson. 2022. “Pytheas: A software package for the automated analysis of RNA sequences and modifications via tandem mass spectrometry.” Nature Communications 13 (1): 2424. https://doi.org/10.1038/s41467-022-30057-5.
Dai, Q., L. S. Zhang, H. L. Sun, K. Pajdzik, L. Yang, C. Ye, C. W. Ju, S. Liu, Y. Wang, Z. Zheng, L. Zhang, B. T. Harada, X. Dou, I. Irkliyenko, X. Feng, W. Zhang, T. Pan, and C. He. 2023. “Quantitative sequencing using BID-seq uncovers abundant pseudouridines in mammalian mRNA at base resolution.” Nature Biotechnology 41 (3): 344–354. https://doi.org/10.1038/s41587-022-01505-w.
Dai, Y., C. B. Qi, Y. Feng, Q. Y. Cheng, F. L. Liu, M. Y. Cheng, B. F. Yuan, and Y. Q. Feng. 2021. “Sensitive and simultaneous determination of uridine thiolation and hydroxylation modifications in eukaryotic RNA by derivatization coupled with mass spectrometry analysis.” Analalytic Chemistry 93 (18): 6938–6946. https://doi.org/10.1021/acs.analchem.0c04630.
Dal Magro, C., P. Keller, A. Kotter, S. Werner, V. Duarte, V. Marchand, M. Ignarski, A. Freiwald, R. U. Müller, C. Dieterich, Y. Motorin, F. Butter, M. Atta, and M. Helm. 2018. “A vastly increased chemical variety of RNA modifications containing a thioacetal structure.” Angewandte Chemie International Edition 57 (26): 7893–7897. https://doi.org/10.1002/anie.201713188.
Dalmay, T., A. Hamilton, S. Rudd, S. Angell, and D. C. Baulcombe. 2000. “An RNA-dependent RNA polymerase gene in Arabidopsis is required for posttranscriptional gene silencing mediated by a transgene but not by a virus.” Cell 101 (5): 543–553. https://doi.org/10.1016/s0092-8674(00)80864-8.
Davis, F. F., and F. W. Allen. 1957. “Ribonucleic acids from yeast which contain a fifth nucleotide.” Journal of Biological Chemistry 227 (2): 907–915. https://doi.org/10.1016/S0021-9258(18)70770-9.
Deamer, D., M. Akeson, and D. Branton. 2016. “Three decades of nanopore sequencing.” Nature Biotechnology 34 (5): 518–524. https://doi.org/10.1038/nbt.3423.
de Crécy-Lagard, V., R. L. Ross, M. Jaroch, V. Marchand, C. Eisenhart, D. Brégeon, Y. Motorin, and P. A. Limbach. 2020. “Survey and validation of tRNA modifications and their corresponding genes in Bacillus subtilis sp subtilis strain 168.” Biomolecules 10 (7). https://doi.org/10.3390/biom10070977.
Dekker, C. 2007. “Solid-state nanopores.” Nature Nanotechnology 2 (4): 209–215. https://doi.org/10.1038/nnano.2007.27.
Demelenne, A., M. J. Gou, G. Nys, C. Parulski, J. Crommen, A. C. Servais, and M. Fillet. 2020. “Evaluation of hydrophilic interaction liquid chromatography, capillary zone electrophoresis and drift tube ion-mobility quadrupole time of flight mass spectrometry for the characterization of phosphodiester and phosphorothioate oligonucleotides.” Journal of Chromatography A 1614: 460716. https://doi.org/10.1016/j.chroma.2019.460716.
Deng, L., J. Kumar, R. Rose, W. McIntyre, and D. Fabris. 2022. “Analyzing RNA posttranscriptional modifications to decipher the epitranscriptomic code.” Mass Spectrometry Reviews: E21798. https://doi.org/10.1002/mas.21798.
Desrosiers, R., K. Friderici, and F. Rottman. 1974. “Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells.” Proceedings of the National Academy of Sciences 71 (10): 3971–3975. https://doi.org/10.1073/pnas.71.10.3971.
Dierks, D., M. A. Garcia-Campos, A. Uzonyi, M. Safra, S. Edelheit, A. Rossi, T. Sideri, R. A. Varier, A. Brandis, Y. Stelzer, F. van Werven, R. Scherz-Shouval, and S. Schwartz. 2021. “Multiplexed profiling facilitates robust m6A quantification at site, gene and sample resolution.” Nature Methods 18 (9): 1060–1067. https://doi.org/10.1038/s41592-021-01242-z.
Dominissini, D., S. Moshitch-Moshkovitz, S. Schwartz, M. Salmon-Divon, L. Ungar, S. Osenberg, K. Cesarkas, J. Jacob-Hirsch, N. Amariglio, M. Kupiec, R. Sorek, and G. Rechavi. 2012. “Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq.” Nature 485 (7397): 201–206. https://doi.org/10.1038/nature11112.
Dominissini, D., S. Nachtergaele, S. Moshitch-Moshkovitz, E. Peer, N. Kol, M. S. Ben-Haim, Q. Dai, A. Di Segni, M. Salmon-Divon, W. C. Clark, G. Zheng, T. Pan, O. Solomon, E. Eyal, V. Hershkovitz, D. Han, L. C. Doré, N. Amariglio, G. Rechavi, and C. He. 2016. “The dynamic N1-methyladenosine methylome in eukaryotic messenger RNA.” Nature 530 (7591): 441–446. https://doi.org/10.1038/nature16998.
Donegan, M., J. M. Nguyen, and M. Gilar. 2022. “Effect of ion-pairing reagent hydrophobicity on liquid chromatography and mass spectrometry analysis of oligonucleotides.” Journal of Chromatography A 1666: 462860. https://doi.org/10.1016/j.chroma.2022.462860.
Eddy, S. R. 2004. “What is a hidden Markov model?” Nature Biotechnology 22 (10): 1315–1316. https://doi.org/10.1038/nbt1004-1315.
Egli, M., and P. S. Pallan. 2010. “Crystallographic studies of chemically modified nucleic acids: A backward glance.” Chemistry & Biodiversity 7 (1): 60–89. https://doi.org/10.1002/cbdv.200900177.
Felden, B., K. Hanawa, J. F. Atkins, H. Himeno, A. Muto, R. F. Gesteland, J. A. McCloskey, and P. F. Crain. 1998. “Presence and location of modified nucleotides in Escherichia coli tmRNA: Structural mimicry with tRNA acceptor branches.” The EMBO Journal 17 (11): 3188–3196. https://doi.org/10.1093/emboj/17.11.3188.
Ferguson, J. M., and M. A. Smith. 2019. “SquiggleKit: A toolkit for manipulating nanopore signal data.” Bioinformatics 35 (24): 5372–5373. https://doi.org/10.1093/bioinformatics/btz586.
Fleming, A. M., N. J. Mathewson, S. A. Howpay Manage, and C. J. Burrows. 2021. “Nanopore dwell time analysis permits sequencing and conformational assignment of pseudouridine in SARS-CoV-2.” ACS Central Science 7 (10): 1707–1717. https://doi.org/10.1021/acscentsci.1c00788.
Fleming, A. M., J. Zhu, V. K. Done, and C. J. Burrows. 2023. “Advantages and challenges associated with bisulfite-assisted nanopore direct RNA sequencing for modifications.” RSC Chemical Biology 4 (11): 952–964. https://doi.org/10.1039/d3cb00081h.
Gao, Y., X. Liu, B. Wu, H. Wang, F. Xi, M. V. Kohnen, A. S. N. Reddy, and L. Gu. 2021. “Quantitative profiling of N6-methyladenosine at single-base resolution in stem-differentiating xylem of Populus trichocarpa using Nanopore direct RNA sequencing.” Genome Biology 22 (1): 22. https://doi.org/10.1186/s13059-020-02241-7.
Garalde, D. R., E. A. Snell, D. Jachimowicz, B. Sipos, J. H. Lloyd, M. Bruce, N. Pantic, T. Admassu, P. James, A. Warland, M. Jordan, J. Ciccone, S. Serra, J. Keenan, S. Martin, L. McNeill, E. J. Wallace, L. Jayasinghe, C. Wright, J. Blasco, S. Young, D. Brocklebank, S. Juul, J. Clarke, A. J. Heron, and D. J. Turner. 2018. “Highly parallel direct RNA sequencing on an array of nanopores.” Nature Methods 15 (3): 201–206. https://doi.org/10.1038/nmeth.4577.
Garcia-Campos, M. A., S. Edelheit, U. Toth, M. Safra, R. Shachar, S. Viukov, R. Winkler, R. Nir, L. Lasman, A. Brandis, J. H. Hanna, W. Rossmanith, and S. Schwartz. 2019. “Deciphering the ‘m6A code’ via antibody-independent quantitative profiling.” Cell 178 (3): 731–747.E16. https://doi.org/10.1016/j.cell.2019.06.013.
Gato, A., M. Catala, C. Tisné, and P. Barraud. 2021. “A method to monitor the introduction of posttranscriptional modifications in tRNAs with NMR spectroscopy.” Methods in Molecular Biology 2298: 307–323. https://doi.org/10.1007/978-1-0716-1374-0_19.
Gerlach, P., H. Malet, S. Cusack, and J. Reguera. 2015. “Structural insights into bunyavirus replication and its regulation by the vRNA promoter.” Cell 161 (6): 1267–1279. https://doi.org/10.1016/j.cell.2015.05.006.
Gilpatrick, T., I. Lee, J. E. Graham, E. Raimondeau, R. Bowen, A. Heron, B. Downs, S. Sukumar, F. J. Sedlazeck, and W. Timp. 2020. “Targeted nanopore sequencing with Cas9-guided adapter ligation.” Nature Biotechnology 38 (4): 433–438. https://doi.org/10.1038/s41587-020-0407-5.
Glasner, H., C. Riml, R. Micura, and K. Breuker. 2017. “Label-free, direct localization and relative quantitation of the RNA nucleobase methylations m6A, m5C, m3U, and m5U by top-down mass spectrometry.” Nucleic Acids Research 45 (13): 8014–8025. https://doi.org/10.1093/nar/gkx470.
Goyal, P., P. V. Krasteva, N. Van Gerven, F. Gubellini, I. Van den Broeck, A. Troupiotis-Tsaïlaki, W. Jonckheere, G. Péhau-Arnaudet, J. S. Pinkner, M. R. Chapman, S. J. Hultgren, S. Howorka, R. Fronzes, and H. Remaut. 2014. “Structural and mechanistic insights into the bacterial amyloid secretion channel CsgG.” Nature 516 (7530): 250–253. https://doi.org/10.1038/nature13768.
Greiner-Stöffele, T., H. H. Foerster, and U. Hahn. 2000. “Ribonuclease T1 cleaves RNA after guanosines within single-stranded gaps of any length.” Nucleosides Nucleotides Nucleic Acids 19 (7): 1101–1109. https://doi.org/10.1080/15257770008035033.
Grosjean, H., G. Keith, and L. Droogmans. 2004. “Detection and quantification of modified nucleotides in RNA using thin-layer chromatography.” In RNA interference, editing, and modification: Methods and protocols, edited by Jonatha M. Gott, Totowa, NJ: Humana Press. Pp. 357–391.
Grozhik, A. V., A. O. Olarerin-George, M. Sindelar, X. Li, S. S. Gross, and S. R. Jaffrey. 2019. “Antibody cross-reactivity accounts for widespread appearance of m1A in 5’UTRs.” Nature Communications 10 (1): 5126. https://doi.org/10.1038/s41467-019-13146-w.
Gupta, R. C., and K. Randerath. 1979. “Rapid print-readout technique for sequencing of RNA’s containing modified nucleotides.” Nucleic Acids Research 6 (11): 3443–3458. https://doi.org/10.1093/nar/6.11.3443.
Guymon, R., S. C. Pomerantz, P. F. Crain, and J. A. McCloskey. 2006. “Influence of phylogeny on posttranscriptional modification of rRNA in thermophilic prokaryotes: The complete modification map of 16S rRNA of Thermusthermophilus.” Biochemistry 45 (15): 4888–4899. https://doi.org/10.1021/bi052579p.
Hagelskamp, F., K. Borland, J. Ramos, A. G. Hendrick, D. Fu, and S. Kellner. 2020. “Broadly applicable oligonucleotide mass spectrometry for the analysis of RNA writers and erasers in vitro.” Nucleic Acids Research 48 (7): E41. https://doi.org/10.1093/nar/gkaa091.
Hagelskamp, F., and S. Kellner. 2021. “Analysis of the epitranscriptome with ion-pairing reagent free oligonucleotide mass spectrometry.” Methods in Enzymology 658: 111–135. https://doi.org/10.1016/bs.mie.2021.06.024.
Hannauer, F., R. Black, A. D. Ray, E. Stulz, G. J. Langley, and S. W. Holman. 2023. “Review of fragmentation of synthetic single-stranded oligonucleotides by tandem mass spectrometry from 2014 to 2022.” Rapid Communications in Mass Spectrometry 37 (17): E9596. https://doi.org/10.1002/rcm.9596.
Hassan, D., D. Acevedo, S. V. Daulatabad, Q. Mir, and S. C. Janga. 2022. “Penguin: A tool for predicting pseudouridine sites in direct RNA nanopore sequencing data.” Methods 203: 478–487. https://doi.org/10.1016/j.ymeth.2022.02.005.
Heiss, M., K. Borland, Y. Yoluç, and S. Kellner. 2021. “Quantification of modified nucleosides in the context of NAIL-MS.” Methods in Molecular Biology 2298: 279–306. https://doi.org/10.1007/978-1-0716-1374-0_18.
Heiss, M., V. F. Reichle, and S. Kellner. 2017. “Observing the fate of tRNA and its modifications by nucleic acid isotope labeling mass spectrometry: NAIL-MS.” RNA Biology 14 (9): 1260–1268. https://doi.org/10.1080/15476286.2017.1325063.
Helm, M., and Y. Motorin. 2017. “Detecting RNA modifications in the epitranscriptome: Predict and validate.” Nature Reviews Genetics 18 (5): 275–291. https://doi.org/10.1038/nrg.2016.169.
Hendra, C., P. N. Pratanwanich, Y. K. Wan, W. S. S. Goh, A. Thiery, and J. Göke. 2022. “Detection of m6A from direct RNA sequencing using a multiple instance learning framework.” Nature Methods 19 (12): 1590–1598. https://doi.org/10.1038/s41592-022-01666-1.
Henley, R. Y., S. Carson, and M. Wanunu. 2016. “Studies of RNA sequence and structure using nanopores.” Progress in Molecular Biology Translational Science 139: 73–99. https://doi.org/10.1016/bs.pmbts.2015.10.020.
Herzog, V. A., B. Reichholf, T. Neumann, P. Rescheneder, P. Bhat, T. R. Burkard, W. Wlotzka, A. von Haeseler, J. Zuber, and S. L. Ameres. 2017. “Thiol-linked alkylation of RNA to assess expression dynamics.” Nature Methods 14 (12): 1198–1204. https://doi.org/10.1038/nmeth.4435.
Hossain, M., and P. A. Limbach. 2007. “Mass spectrometry-based detection of transfer RNAs by their signature endonuclease digestion products.” RNA 13 (2): 295–303. https://doi.org/10.1261/rna.272507.
Houser, W. M., A. Butterer, B. Addepalli, and P. A. Limbach. 2015. “Combining recombinant ribonuclease U2 and protein phosphatase for RNA modification mapping by liquid chromatography-mass spectrometry.” Analytical Biochemistry 478: 52–58. https://doi.org/10.1016/j.ab.2015.03.016.
Hu, L., S. Liu, Y. Peng, R. Ge, R. Su, C. Senevirathne, B. T. Harada, Q. Dai, J. Wei, L. Zhang, Z. Hao, L. Luo, H. Wang, Y. Wang, M. Luo, M. Chen, J. Chen, and C. He. 2022. “m6A RNA modifications are measured at single-base resolution across the mammalian transcriptome.” Nature Biotechnology 40 (8): 1210–1219. https://doi.org/10.1038/s41587-022-01243-z.
Huang, G., F. Zhang, D. Xie, Y. Ma, P. Wang, G. Cao, L. Chen, S. Lin, Z. Zhao, and Z. Cai. 2023. “High-throughput profiling of RNA modifications by ultra-performance liquid chromatography coupled to complementary mass spectrometry: Methods, quality control, and applications.” Talanta 263: 124697. https://doi.org/10.1016/j.talanta.2023.124697.
Huang, S., W. Zhang, C. D. Katanski, D. Dersh, Q. Dai, K. Lolans, J. Yewdell, A. M. Eren, and T. Pan. 2021. “Interferon inducible pseudouridine modification in human mRNA by quantitative nanopore profiling.” Genome Biology 22 (1): 330. https://doi.org/10.1186/s13059-021-02557-y.
Ip, C. L. C., M. Loose, J. R. Tyson, M. de Cesare, B. L. Brown, M. Jain, R. M. Leggett, D. A. Eccles, V. Zalunin, J. M. Urban, P. Piazza, R. J. Bowden, B. Paten, S. Mwaigwisya, E. M. Batty, J. T. Simpson, T. P. Snutch, E. Birney, D. Buck, S. Goodwin, H. J. Jansen, J. O’Grady, and H. E. Olsen. 2015. “MinION analysis and reference consortium: Phase 1 data release and analysis.” F1000Research 4: 1075. https://doi.org/10.12688/f1000research.7201.1.
Jackson, R. W., C. M. Smathers, and A. R. Robart. 2023. “General strategies for RNA x-ray crystallography.” Molecules 28 (5). https://doi.org/10.3390/molecules28052111.
Jain, M., H. E. Olsen, B. Paten, and M. Akeson. 2016. “The Oxford nanopore MinION: Delivery of nanopore sequencing to the genomics community.” Genome Biology 17 (1): 239. https://doi.org/10.1186/s13059-016-1103-0.
Janssen, K. A., Y. Xie, M. C. Kramer, B. D. Gregory, and B. A. Garcia. 2022. “Data-independent acquisition for the detection of mononucleoside RNA modifications by mass spectrometry.” Journal of the American Society for Mass Spectrometry 33 (5): 885–893. https://doi.org/10.1021/jasms.2c00065.
Jenjaroenpun, P., T. Wongsurawat, T. Wadley, T. Wassenaar, L. Jun, Q. Dai, V. Wanchai, N. Akel, a. Jamshidi, A. Franco, G. Boysen, M. Jennings, D. Ussery, C. He, and I. Nookaew. 2020. “Decoding the epitranscriptional landscape from native RNA sequences.” Nucleic Acids Research 49. https://doi.org/10.1093/nar/gkaa620.
Jiang, T., N. Yu, J. Kim, J. R. Murgo, M. Kissai, K. Ravichandran, E. J. Miracco, V. Presnyak, and S. Hua. 2019. “Oligonucleotide sequence mapping of large therapeutic mRNAs via parallel ribonuclease digestions and LC-MS/MS.” Analytical Chemistry 91 (13): 8500–8506. https://doi.org/10.1021/acs.analchem.9b01664.
Jones, J. D., K. M. Simcox, R. T. Kennedy, and K. S. Koutmou. 2023. “Direct sequencing of total Saccharomyces cerevisiae tRNAs by LC-MS/MS.” RNA 29 (8): 1201–1214. https://doi.org/10.1261/rna.079656.123.
Jora, M., K. Borland, S. Abernathy, R. Zhao, M. Kelley, S. Kellner, B. Addepalli, and P. A. Limbach. 2021. “Chemical amination/imination of carbonothiolated nucleosides during RNA hydrolysis.” Angewandte Chemie International Edition 60 (8): 3961–3966. https://doi.org/10.1002/anie.202010793.
Jora, M., A. P. Burns, R. L. Ross, P. A. Lobue, R. Zhao, C. M. Palumbo, P. A. Beal, B. Addepalli, and P. A. Limbach. 2018. “Differentiating positional isomers of nucleoside modifications by higher-energy collisional dissociation mass spectrometry (HCD MS).” Journal of the American Society for Mass Spectrometry 29 (8): 1745–1756. https://doi.org/10.1007/s13361-018-1999-6.
Jora, M., D. Corcoran, G. G. Parungao, P. A. Lobue, L. F. L. Oliveira, G. Stan, B. Addepalli, and P. A. Limbach. 2022. “Higher-energy collisional dissociation mass spectral networks for the rapid, semi-automated characterization of known and unknown ribonucleoside modifications.” Analytical Chemistry 94 (40): 13958–13967. https://doi.org/10.1021/acs.analchem.2c03172.
Jora, M., P. A. Lobue, R. L. Ross, B. Williams, and B. Addepalli. 2019. “Detection of ribonucleoside modifications by liquid chromatography coupled with mass spectrometry.” Biochimica Biophysica Acta Gene Regulatory Mechanisms 1862 (3): 280–290. https://doi.org/10.1016/j.bbagrm.2018.10.012.
Kadumuri, R. V., and S. C. Janga. 2018. “Epitranscriptomic code and its alterations in human disease.” Trends in Molecular Medicine 24 (10): 886–903. https://doi.org/10.1016/j.molmed.2018.07.010.
Kaiser, S., S. R. Byrne, G. Ammann, P. Asadi Atoi, K. Borland, R. Brecheisen, M. S. DeMott, T. Gehrke, F. Hagelskamp, M. Heiss, Y. Yoluç, L. Liu, Q. Zhang, P. C. Dedon, B. Cao, and S. Kellner. 2021. “Strategies to avoid artifacts in mass spectrometry-based epitranscriptome analyses.” Angewandte Chemie International Edition 60 (44): 23885–23893. https://doi.org/10.1002/anie.202106215.
Kamath, G. M., I. Shomorony, F. Xia, T. A. Courtade, and D. N. Tse. 2017. “HINGE: Long-read assembly achieves optimal repeat resolution.” Genome Research 27 (5): 747–756. https://doi.org/10.1101/gr.216465.116.
Kang, B. I., K. Miyauchi, M. Matuszewski, G. S. D’Almeida, M. A. T. Rubio, J. D. Alfonzo, K. Inoue, Y. Sakaguchi, T. Suzuki, E. Sochacka, and T. Suzuki. 2017. “Identification of 2-methylthio cyclic N6-threonylcarbamoyladenosine (ms2ct6A) as a novel RNA modification at position 37 of tRNAs.” Nucleic Acids Research 45 (4): 2124–2136. https://doi.org/10.1093/nar/gkw1120.
Kasimova, M. A., E. Lindahl, and L. Delemotte. 2018. “Determining the molecular basis of voltage sensitivity in membrane proteins.” Journal of General Physiology 150 (10): 1444–1458. https://doi.org/10.1085/jgp.201812086.
Ke, S., E. A. Alemu, C. Mertens, E. C. Gantman, J. J. Fak, A. Mele, B. Haripal, I. Zucker-Scharff, M. J. Moore, C. Y. Park, C. B. Vågbø, A. Kusśnierczyk, A. Klungland, J. E. Darnell, Jr., and R. B. Darnell. 2015. “A majority of m6A residues are in the last exons, allowing the potential for 3’ UTR regulation.” Genes & Development 29 (19): 2037–2053. https://doi.org/10.1101/gad.269415.115.
Keller, M. W., B. L. Rambo-Martin, M. M. Wilson, C. A. Ridenour, S. S. Shepard, T. J. Stark, E. B. Neuhaus, V. G. Dugan, D. E. Wentworth, and J. R. Barnes. 2018. “Direct RNA sequencing of the coding complete influenza A virus genome.” Scientific Reports 8 (1). https://doi.org/10.1038/s41598-018-32615-8.
Kellner, S., J. Burhenne, and M. Helm. 2010. “Detection of RNA modifications.” RNA biology 7 (2): 237–247. https://doi.org/10.4161/rna.7.2.11468.
Kellner, S., J. Neumann, D. Rosenkranz, S. Lebedeva, R. F. Ketting, H. Zischler, D. Schneider, and M. Helm. 2014. “Profiling of RNA modifications by multiplexed stable isotope labelling.” Chemical Communications 50 (26): 3516–3518. https://doi.org/10.1039/c3cc49114e.
Kenderdine, T., R. Nemati, A. Baker, M. Palmer, J. Ujma, M. FitzGibbon, L. Deng, M. Royzen, J. Langridge, and D. Fabris. 2020. “High-resolution ion mobility spectrometry-mass spectrometry of isomeric/isobaric ribonucleotide variants.” Journal of Mass Spectrometry 55 (2): E4465. https://doi.org/10.1002/jms.4465.
Khoddami, V., A. Yerra, T. L. Mosbruger, A. M. Fleming, C. J. Burrows, and B. R. Cairns. 2019. “Transcriptome-wide profiling of multiple RNA modifications simultaneously at single-base resolution.” Proceedings of the National Academy of Sciences 116 (14): 6784–6789. https://doi.org/10.1073/pnas.1817334116.
Kimura, S., P. C. Dedon, and M. K. Waldor. 2020. “Comparative tRNA sequencing and RNA mass spectrometry for surveying tRNA modifications.” Nature Chemical Biology 16 (9): 964–972. https://doi.org/10.1038/s41589-020-0558-1.
Kimura, S., V. Srisuknimit, and M. K. Waldor. 2020. “Probing the diversity and regulation of tRNA modifications.” Current Opinion in Microbiology 57: 41–48. https://doi.org/10.1016/j.mib.2020.06.005.
Kong, Y., E. A. Mead, and G. Fang. 2023. “Navigating the pitfalls of mapping DNA and RNA modifications.” Nature Reviews Genetics 24 (6): 363–381. https://doi.org/10.1038/s41576-022-00559-5.
Koonchanok, R., S. V. Daulatabad, Q. Mir, K. Reda, and S. C. Janga. 2021. “Sequoia: An interactive visual analytics platform for interpretation and feature extraction from nanopore sequencing datasets.” BMC Genomics 22 (1): 513. https://doi.org/10.1186/s12864-021-07791-z.
Koonchanok, R., S. V. Daulatabad, K. Reda, and S. C. Janga. 2023. “Sequoia: A framework for visual analysis of RNA modifications from direct RNA sequencing data.” Methods in Molecular Biology 2624: 127–138. https://doi.org/10.1007/978-1-0716-2962-8_9.
Koren, S., B. P. Walenz, K. Berlin, J. R. Miller, N. H. Bergman, and A. M. Phillippy. 2017. “Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation.” Genome Research 27 (5): 722–736. https://doi.org/10.1101/gr.215087.116.
Kowalak, J. A., E. Bruenger, and J. A. McCloskey. 1995. “Posttranscriptional modification of the central loop of domain V in Escherichia coli 23 S ribosomal RNA.” Journal of Biological Chemistry 270 (30): 17758–17764. https://doi.org/10.1074/jbc.270.30.17758.
Kowalak, J. A., S. C. Pomerantz, P. F. Crain, and J. A. McCloskey. 1993. “A novel method for the determination of posttranscriptional modification in RNA by mass spectrometry.” Nucleic Acids Research 21 (19): 4577–4585. https://doi.org/10.1093/nar/21.19.4577.
Lander, E. S., L. M. Linton, B. Birren, C. Nusbaum, M. C. Zody, J. Baldwin, K. Devon, K. Dewar, M. Doyle, W. FitzHugh, R. Funke, D. Gage, K. Harris, A. Heaford, J. Howland, L. Kann, J. Lehoczky, R. LeVine, P. McEwan, K. McKernan, J. Meldrim, J. P. Mesirov, C. Miranda, W. Morris, J. Naylor, C. Raymond, M. Rosetti, R. Santos, A. Sheridan, C. Sougnez, Y. Stange-Thomann, N. Stojanovic, A. Subramanian, D. Wyman, J. Rogers, J. Sulston, R. Ainscough, S. Beck, D. Bentley, J. Burton, C. Clee, N. Carter, A. Coulson, R. Deadman, P. Deloukas, A. Dunham, I. Dunham, R. Durbin, L. French, D. Grafham, S. Gregory, T. Hubbard, S. Humphray, A. Hunt, M. Jones, C. Lloyd, A. McMurray, L. Matthews, S. Mercer, S. Milne, J. C. Mullikin, A. Mungall, R. Plumb, M. Ross, R. Shownkeen, S. Sims, R. H. Waterston, R. K. Wilson, L. W. Hillier, J. D. McPherson, M. A. Marra, E. R. Mardis, L. A. Fulton, A. T. Chinwalla, K. H. Pepin, W. R. Gish, S. L. Chissoe, M. C. Wendl, K. D. Delehaunty, T. L. Miner, A. Delehaunty, J. B. Kramer, L. L. Cook, R. S. Fulton, D. L. Johnson, P. J. Minx, S. W. Clifton, T. Hawkins, E. Branscomb, P. Predki, P. Richardson, S. Wenning, T. Slezak, N. Doggett, J. F. Cheng, A. Olsen, S. Lucas, C. Elkin, E. Uberbacher, M. Frazier, R. A. Gibbs, D. M. Muzny, S. E. Scherer, J. B. Bouck, E. J. Sodergren, K. C. Worley, C. M. Rives, J. H. Gorrell, M. L. Metzker, S. L. Naylor, R. S. Kucherlapati, D. L. Nelson, G. M. Weinstock, Y. Sakaki, A. Fujiyama, M. Hattori, T. Yada, A. Toyoda, T. Itoh, C. Kawagoe, H. Watanabe, Y. Totoki, T. Taylor, J. Weissenbach, R. Heilig, W. Saurin, F. Artiguenave, P. Brottier, T. Bruls, E. Pelletier, C. Robert, P. Wincker, D. R. Smith, L. Doucette-Stamm, M. Rubenfield, K. Weinstock, H. M. Lee, J. Dubois, A. Rosenthal, M. Platzer, G. Nyakatura, S. Taudien, A. Rump, H. Yang, J. Yu, J. Wang, G. Huang, J. Gu, L. Hood, L. Rowen, A. Madan, S. Qin, R. W. Davis, N. A. Federspiel, A. P. Abola, M. J. Proctor, R. M. Myers, J. Schmutz, M. Dickson, J. Grimwood, D. R. Cox, M. V. Olson, R. Kaul, C. Raymond, N. Shimizu, K. Kawasaki, S. Minoshima, G. A. Evans, M. Athanasiou, R. Schultz, B. A. Roe, F. Chen, H. Pan, J. Ramser, H. Lehrach, R. Reinhardt, W. R. McCombie, M. de la Bastide, N. Dedhia, H. Blöcker, K. Hornischer, G. Nordsiek, R. Agarwala, L. Aravind, J. A. Bailey, A. Bateman, S. Batzoglou, E. Birney, P. Bork, D. G. Brown, C. B. Burge, L. Cerutti, H. C. Chen, D. Church, M. Clamp, R. R. Copley, T. Doerks, S. R. Eddy, E. E. Eichler, T. S. Furey, J. Galagan, J. G. Gilbert, C. Harmon, Y. Hayashizaki, D. Haussler, H. Hermjakob, K. Hokamp, W. Jang, L. S. Johnson, T. A. Jones, S. Kasif, A. Kaspryzk, S. Kennedy, W. J. Kent, P. Kitts, E. V. Koonin, I. Korf, D. Kulp, D. Lancet, T. M. Lowe, A. McLysaght, T. Mikkelsen, J. V. Moran, N. Mulder, V. J. Pollara, C. P. Ponting, G. Schuler, J. Schultz, G. Slater, A. F. Smit, E. Stupka, J. Szustakowki, D. Thierry-Mieg, J. Thierry-Mieg, L. Wagner, J. Wallis, R. Wheeler, A. Williams, Y. I. Wolf, K. H. Wolfe, S. P. Yang, R. F. Yeh, F. Collins, M. S. Guyer, J. Peterson, A. Felsenfeld, K. A. Wetterstrand, A. Patrinos, M. J. Morgan, P. de Jong, J. J. Catanese, K. Osoegawa, H. Shizuya, S. Choi, Y. J. Chen, and J. Szustakowki. 2001. “Initial sequencing and analysis of the human genome.” Nature 409 (6822): 860–921. https://doi.org/10.1038/35057062.
Leger, A., P. P. Amaral, L. Pandolfini, C. Capitanchik, F. Capraro, V. Miano, V. Migliori, P. Toolan-Kerr, T. Sideri, A. J. Enright, K. Tzelepis, F. J. van Werven, N. M. Luscombe, I. Barbieri, J. Ule, T. Fitzgerald, E. Birney, T. Leonardi, and T. Kouzarides. 2021. “RNA modifications detection by comparative Nanopore direct RNA sequencing.” Nature Communications 12 (1): 7198. https://doi.org/10.1038/s41467-021-27393-3.
Leggett, R. M., D. Heavens, M. Caccamo, M. D. Clark, and R. P. Davey. 2015. “NanoOK: Multi-reference alignment analysis of nanopore sequencing data, quality and error profiles.” Bioinformatics 32 (1): 142–144. https://doi.org/10.1093/bioinformatics/btv540.
Lewis, C., Pan, T. and Kalsotra, A. RNA modifications and structures cooperate to guide RNA–protein interactions. Nature Reviews Molecular Cell Biology 18: 202–210. https://doi.org/10.1038/nrm.2016.163
Li, F., X. Guo, P. Jin, J. Chen, D. Xiang, J. Song, and L. J. M. Coin. 2021. “Porpoise: A new approach for accurate prediction of RNA pseudouridine sites.” Briefings in Bioinformatics 22 (6). https://doi.org/10.1093/bib/bbab245.
Li, J., Z. Chen, F. Chen, G. Xie, Y. Ling, Y. Peng, Y. Lin, N. Luo, C. M. Chiang, and H. Wang. 2020. “Targeted mRNA demethylation using an engineered dCas13b-ALKBH5 fusion protein.” Nucleic Acids Research 48 (10): 5684–5694. https://doi.org/10.1093/nar/gkaa269.
Li, X., P. Zhu, S. Ma, J. Song, J. Bai, F. Sun, and C. Yi. 2015. “Chemical pulldown reveals dynamic pseudouridylation of the mammalian transcriptome.” Nature Chemical Biology 11 (8): 592–597. https://doi.org/10.1038/nchembio.1836.
Lin, S., Q. Liu, V. S. Lelyveld, J. Choe, J. W. Szostak, and R. I. Gregory. 2018. “Mettl1/Wdr4-mediated m7G tRNA methylome is required for normal mRNA translation and embryonic stem cell self-Renewal and Differentiation.” Molecular Cell 71 (2): 244-255.E5. https://doi.org/10.1016/j.molcel.2018.06.001.
Lin, X., Q. Zhang, Y. Qin, Q. Zhong, D. Lv, X. Wu, P. Fu, and H. Lin. 2022. “Potential misidentification of natural isomers and mass-analogs of modified nucleosides by liquid chromatography–triple quadrupole mass spectrometry.” Genes 13 (5): 878.
Linder, B., A. V. Grozhik, A. O. Olarerin-George, C. Meydan, C. E. Mason, and S. R. Jaffrey. 2015. “Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome.” Nature Methods 12 (8): 767–772. https://doi.org/10.1038/nmeth.3453.
Liu, C., H. Sun, Y. Yi, W. Shen, K. Li, Y. Xiao, F. Li, Y. Li, Y. Hou, B. Lu, W. Liu, H. Meng, J. Peng, C. Yi, and J. Wang. 2023. “Absolute quantification of single-base m6A methylation in the mammalian transcriptome using GLORI.” Nature Biotechnology 41 (3): 355–366. https://doi.org/10.1038/s41587-022-01487-9.
Liu, H., O. Begik, M. C. Lucas, J. M. Ramirez, C. E. Mason, D. Wiener, S. Schwartz, J. S. Mattick, M. A. Smith, and E. M. Novoa. 2019a. “Accurate detection of m6A RNA modifications in native RNA sequences.” Nature Communications 10 (1): 4079. https://doi.org/10.1038/s41467-019-11713-9.
Liu, N., Q. Dai, G. Zheng, C. He, M. Parisien, and T. Pan. 2015. “N6-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions.” Nature 518 (7540): 560–564. https://doi.org/10.1038/nature14234.
Liu, R., L. Ou, B. Sheng, P. Hao, P. Li, X. Yang, G. Xue, L. Zhu, Y. Luo, P. Zhang, P. Yang, H. Li, and D. D. Feng. 2022. “Mixed-weight neural bagging for detecting m6A modifications in SARS-CoV-2 RNA sequencing.” IEEE Transactions on Biomedical Engineering 69 (8): 2557–2568. https://doi.org/10.1109/tbme.2022.3150420.
Liu, X. M., J. Zhou, Y. Mao, Q. Ji, and S. B. Qian. 2019b. “Programmable RNA N6-methyladenosine editing by CRISPRCas9 conjugates.” Nature Chemical Biology 15 (9): 865–871. https://doi.org/10.1038/s41589-019-0327-1.
Liu, Y., Y. Rodriguez, R. L. Ross, R. Zhao, J. A. Watts, C. Grunseich, A. Bruzel, D. Li, J. T. Burdick, R. Prasad, R. J. Crouch, P. A. Limbach, S. H. Wilson, and V. G. Cheung. 2020. “RNA abasic sites in yeast and human cells.” Proceedings of the National Academy of Sciences 117 (34): 20689–20695. https://doi.org/10.1073/pnas.2011511117.
Loman, N. J., J. Quick, and J. T. Simpson. 2015. “A complete bacterial genome assembled de novo using only nanopore sequencing data.” Nature Methods 12 (8): 733–735. https://doi.org/10.1038/nmeth.3444.
Loman, N. J., and A. R. Quinlan. 2014. “Poretools: A toolkit for analyzing nanopore sequence data.” Bioinformatics 30 (23): 3399–3401. https://doi.org/10.1093/bioinformatics/btu555.
Lorenz, D. A., S. Sathe, J. M. Einstein, and G. W. Yeo. 2020. “Direct RNA sequencing enables m6A detection in endogenous transcript isoforms at base-specific resolution.” RNA 26 (1): 19–28. https://doi.org/10.1261/rna.072785.119.
Macias, L. A., S. P. Garcia, K. M. Back, Y. Wu, G. H. Johnson, S. Kathiresan, A. M. Bellinger, E. Rohde, M. A. Freitas, and J. A. Madsen. 2023. “Spacer fidelity assessments of guide RNA by top-down mass spectrometry.” ACS Central Science 9 (7): 1437–1452. https://doi.org/10.1021/acscentsci.3c00289.
MacKenzie, M., and C. Argyropoulos. 2023. “An introduction to nanopore sequencing: Past, present, and future considerations.” Micromachines 14 (2): 459. https://doi.org/10.3390/mi14020459.
Maier, K. C., S. Gressel, P. Cramer, and B. Schwalb. 2020. “Native molecule sequencing by nano-ID reveals synthesis and stability of RNA isoforms.” Genome Research 30 (9): 1332–1344. https://doi.org/10.1101/gr.257857.119.
Marchand, V., F. Blanloeil-Oillo, M. Helm, and Y. Motorin. 2016. “Illumina-based RiboMethSeq approach for mapping of 2’-O-Me residues in RNA.” Nucleic Acids Research 44 (16): E135. https://doi.org/10.1093/nar/gkw547.
Marchand, V., F. Pichot, P. Neybecker, L. Ayadi, V. Bourguignon-Igel, L. Wacheul, D. L. J. Lafontaine, A. Pinzano, M. Helm, and Y. Motorin. 2020. “HydraPsiSeq: A method for systematic and quantitative mapping of pseudouridines in RNA.” Nucleic Acids Research 48 (19): E110. https://doi.org/10.1093/nar/gkaa769.
Marx, V. 2021. “Long road to long-read assembly.” Nature Methods 18 (2): 125–129. https://doi.org/10.1038/s41592-021-01057-y.
Matuszewski, M., J. Wojciechowski, K. Miyauchi, Z. Gdaniec, W. M. Wolf, T. Suzuki, and E. Sochacka. 2017. “A hydantoin isoform of cyclic N6-threonylcarbamoyladenosine (ct6A) is present in tRNAs.” Nucleic Acids Research 45 (4): 2137–2149. https://doi.org/10.1093/nar/gkw1189.
Mayer, S. F., C. Cao, and M. Dal Peraro. 2022. “Biological nanopores for single-molecule sensing.” iScience 25 (4): 104145. https://doi.org/10.1016/j.isci.2022.104145.
McCown, P. J., A. Ruszkowska, C. N. Kunkler, K. Breger, J. P. Hulewicz, M. C. Wang, N. A. Springer, and J. A. Brown. 2020. “Naturally occurring modified ribonucleosides.” WIREs RNA 11 (5): E1595. https://doi.org/10.1002/wrna.1595.
McMahon, M. 2023. “Artificial Intelligence for Sequence and Structure Recognition.” ReviR Therapeutics, Inc. Presentation to the National Academies. https://www.nationalacademies.org/event/03-13-2023/toward-sequencing-and-mapping-of-rna-modifications-a-workshop.
McLuckey, S. A., G. J. Van Berkel, and G. L. Glish. 1992. “Tandem mass spectrometry of small, multiply charged oligonucleotides.” Journal of the American Society for Mass Spectrometry 3 (1): 60–70. https://doi.org/10.1016/1044-0305(92)85019-g.
Meyer, K. D. 2019. “DART-seq: An antibody-free method for global m6A detection.” Nature Methods 16 (12): 1275–1280. https://doi.org/10.1038/s41592-019-0570-0.
Meyer, K. D., Y. Saletore, P. Zumbo, O. Elemento, C. E. Mason, and S. R. Jaffrey. 2012. “Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons.” Cell 149 (7): 1635–1646. https://doi.org/10.1016/j.cell.2012.05.003.
Minchin, S., and J. Lodge. 2019. “Understanding biochemistry: Structure and function of nucleic acids.” Essays in Biochemistry 63 (4): 433–456. https://doi.org/10.1042/ebc20180038.
Mola, P. 2023. “Digitizing Biology.” Roswell Biotechnologies. Presentation to the National Academies. https://www.youtube.com/watch?v=5FSMtqUyOFM.
Molinie, B., J. Wang, K. S. Lim, R. Hillebrand, Z. X. Lu, N. Van Wittenberghe, B. D. Howard, K. Daneshvar, A. C. Mullen, P. Dedon, Y. Xing, and C. C. Giallourakis. 2016. “m6A-LAIC-seq reveals the census and complexity of the m6A epitranscriptome.” Nature Methods 13 (8): 692–698. https://doi.org/10.1038/nmeth.3898.
Napieralski, A., and R. Nowak. 2022. “Basecalling using joint raw and event nanopore data sequence-to sequence processing.” Sensors 22 (6): 2275. https://doi.org/10.3390/s22062275.
NASEM (National Academies of Sciences, Engineering, and Medicine). 2023. Toward sequencing and mapping of RNA modifications: Proceedings of a workshop–in brief. Edited by Steven Moss and Michael Zierler. Washington, DC: The National Academies Press. https://nap.nationalacademies.org/read/27149/chapter/1 (accessed March 11, 2024).
Neidle, S., and H. M. Berman. 1983. “X-ray crystallographic studies of nucleic acids and nucleic acid-drug complexes.” Progress in Biophysics and Molecular Biology 41 (2): 43–66. https://doi.org/10.1016/0079-6107(83)90025-1.
Okada, S., H. Ueda, Y. Noda, and T. Suzuki. 2019. “Transcriptome-wide identification of A-to-I RNA editing sites using ICE-seq.” Methods 156: 66–78. https://doi.org/10.1016/j.ymeth.2018.12.007.
Orts, J., and A. D. Gossert. 2018. “Structure determination of protein-ligand complexes by NMR in solution.” Methods 138-139: 3–25. https://doi.org/10.1016/j.ymeth.2018.01.019.
Pandolfini, L., I. Barbieri, A. J. Bannister, A. Hendrick, B. Andrews, N. Webster, P. Murat, P. Mach, R. Brandi, S. C. Robson, V. Migliori, A. Alendar, M. d’Onofrio, S. Balasubramanian, and T. Kouzarides. 2019. “METTL1 promotes let-7 MicroRNA processing via m7G methylation.” Molecular Cell 74 (6): 1278–1290.E9. https://doi.org/10.1016/j.molcel.2019.03.040.
Pellegrino, S., K. C. Dent, T. Spikes, and A. J. Warren. 2023. “Cryo-EM reconstruction of the human 40S ribosomal subunit at 2.15 Å resolution.” Nucleic Acids Research 51 (8): 4043–4054. https://doi.org/10.1093/nar/gkad194.
Peters-Clarke, T. M., Q. Quan, D. R. Brademan, A. S. Hebert, M. S. Westphall, and J. J. Coon. 2020. “Ribonucleic acid sequence characterization by negative electron transfer dissociation mass spectrometry.” Analalytical Chemistry 92 (6): 4436–4444. https://doi.org/10.1021/acs.analchem.9b05388.
Pomerantz, S. C., J. A. Kowalak, and J. A. McCloskey. 1993. “Determination of oligonucleotide composition from mass spectrometrically measured molecular weight.” Journal of the American Society for Mass Spectrometry 4 (3): 204–209. https://doi.org/10.1016/1044-0305(93)85082-9.
Pratanwanich, P. N., F. Yao, Y. Chen, C. W. Q. Koh, Y. K. Wan, C. Hendra, P. Poon, Y. T. Goh, P. M. L. Yap, J. Y. Chooi, W. J. Chng, S. B. Ng, A. Thiery, W. S. S. Goh, and J. Göke. 2021. “Identification of differential RNA modifications from nanopore direct RNA sequencing with xPore.” Nature Biotechnology 39 (11): 1394–1402. https://doi.org/10.1038/s41587-021-00949-w.
Prats-Ejarque, G., L. Lu, V. A. Salazar, M. Moussaoui, and E. Boix. 2019. “Evolutionary trends in RNA base selectivity within the RNase A superfamily.” Frontiers in Pharmacology 10: 1170. https://doi.org/10.3389/fphar.2019.01170.
Punthambaker, S. 2022. “Detection of modified RNA with an engineered nanopore.” Nature Nanotechnology 17 (10): 1044–1045. https://doi.org/10.1038/s41565-022-01210-4.
Puri, P., C. Wetzel, P. Saffert, K. W. Gaston, S. P. Russell, J. A. Cordero Varela, P. van der Vlies, G. Zhang, P. A. Limbach, Z. Ignatova, and B. Poolman. 2014. “Systematic identification of tRNAome and its dynamics in Lactococcus lactis.” Molecular Microbiology 93 (5): 944–956. https://doi.org/10.1111/mmi.12710.
Qin, H., L. Ou, J. Gao, L. Chen, J. W. Wang, P. Hao, and X. Li. 2022. “DENA: Training an authentic neural network model using Nanopore sequencing data of Arabidopsis transcripts for detection and quantification of N6-methyladenosine on RNA.” Genome Biology 23 (1): 25. https://doi.org/10.1186/s13059-021-02598-3.
Rang, F. J., W. P. Kloosterman, and J. de Ridder. 2018. “From squiggle to basepair: computational approaches for improving nanopore sequencing read accuracy.” Genome Biology 19 (1): 90. https://doi.org/10.1186/s13059-018-1462-9.
Ramakrishnan, V. 2002. “Ribosome structure and the mechanism of translation.” Cell 108 (4): 557–572. https://doi.org/10.1016/s0092-8674(02)00619-0.
Ramasamy, S., S. Mishra, S. Sharma, S. S. Parimalam, T. Vaijayanthi, Y. Fujita, B. Kovi, H. Sugiyama, and G. N. Pandian. 2022. “An informatics approach to distinguish RNA modifications in nanopore direct RNA sequencing.” Genomics 114 (3): 110372. https://doi.org/10.1016/j.ygeno.2022.110372.
Reich, D. P., and B. L. Bass. 2019. “Mapping the dsRNA world.” Cold Spring Harbor Perspectives in Biology 11 (3). https://doi.org/10.1101/cshperspect.a035352.
Riml, C., T. Amort, D. Rieder, C. Gasser, A. Lusser, and R. Micura. 2017. “Osmium-mediated transformation of 4-thiouri-dine to cytidine as key to study RNA dynamics by sequencing.” Angewandte Chemie International Edition 56 (43): 13479–13483. https://doi.org/10.1002/anie.201707465.
Roberts, J. T., A. M. Porman, and A. M. Johnson. 2021. “Identification of m6A residues at single-nucleotide resolution using eCLIP and an accessible custom analysis pipeline.” RNA 27 (4): 527–541. https://doi.org/10.1261/rna.078543.120.
Roth, S. H., E. Y. Levanon, and E. Eisenberg. 2019. “Genome-wide quantification of ADAR adenosine-to-inosine RNA editing activity.” Nature Methods 16 (11): 1131–1138. https://doi.org/10.1038/s41592-019-0610-9.
Ruiz-Arroyo, V. M., R. Raj, K. Babu, O. Onolbaatar, P. H. Roberts, and Y. Nam. 2023. “Structures and mechanisms of tRNA methylation by METTL1-WDR4.” Nature 613 (7943): 383–390. https://doi.org/10.1038/s41586-022-05565-5.
Sample, P. J., K. W. Gaston, J. D. Alfonzo, and P. A. Limbach. 2015. “RoboOligo: Software for mass spectrometry data to support manual and de novo sequencing of post-transcriptionally modified ribonucleic acids.” Nucleic Acids Research 43 (10): E64. https://doi.org/10.1093/nar/gkv145.
Santos, I. C., M. Lanzillotti, I. Shilov, M. Basanta-Sanchez, A. Roushan, R. Lawler, W. Tang, M. Bern, and J. S. Brodbelt. 2022. “Ultraviolet photodissociation and activated electron photodetachment mass spectrometry for top-down sequencing of modified oligoribonucleotides.” Journal of the American Society for Mass Spectrometry 33 (3): 510–520. https://doi.org/10.1021/jasms.1c00340.
Sarkar, A., W. Gasperi, U. Begley, S. Nevins, S. M. Huber, P. C. Dedon, and T. J. Begley. 2021. “Detecting the epitranscriptome.” WIREs RNA 12 (6): E1663. https://doi.org/10.1002/wrna.1663.
Sas-Chen, A., J. M. Thomas, D. Matzov, M. Taoka, K. D. Nance, R. Nir, K. M. Bryson, R. Shachar, G. L. S. Liman, B. W. Burkhart, S. T. Gamage, Y. Nobe, C. A. Briney, M. J. Levy, R. T. Fuchs, G. B. Robb, J. Hartmann, S. Sharma, Q. Lin, L. Florens, M. P. Washburn, T. Isobe, T. J. Santangelo, M. Shalev-Benami, J. L. Meier, and S. Schwartz. 2020. “Dynamic RNA acetylation revealed by quantitative cross-evolutionary mapping.” Nature 583 (7817): 638–643. https://doi.org/10.1038/s41586-020-2418-2.
Schaefer, M., T. Pollex, K. Hanna, and F. Lyko. 2009. “RNA cytosine methylation analysis by bisulfite sequencing.” Nucleic Acids Research 37 (2): E12. https://doi.org/10.1093/nar/gkn954.
Schofield, J. A., E. E. Duffy, L. Kiefer, M. C. Sullivan, and M. D. Simon. 2018. “TimeLapse-seq: Adding a temporal dimension to RNA sequencing through nucleoside recoding.” Nature Methods 15 (3): 221–225. https://doi.org/10.1038/nmeth.4582.
Schwartz, S., D. A. Bernstein, M. R. Mumbach, M. Jovanovic, R. H. Herbst, B. X. León-Ricardo, J. M. Engreitz, M. Gutt-man, R. Satija, E. S. Lander, G. Fink, and A. Regev. 2014. “Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA.” Cell 159 (1): 148–162. https://doi.org/10.1016/j.cell.2014.08.028.
Semeraro, R., and A. Magi. 2019. “PyPore: A python toolbox for nanopore sequencing data handling.” Bioinformatics 35 (21): 4445–4447. https://doi.org/10.1093/bioinformatics/btz269.
Shafin, K., T. Pesout, R. Lorig-Roach, M. Haukness, H. E. Olsen, C. Bosworth, J. Armstrong, K. Tigyi, N. Maurer, S. Koren, F. J. Sedlazeck, T. Marschall, S. Mayes, V. Costa, J. M. Zook, K. J. Liu, D. Kilburn, M. Sorensen, K. M. Munson, M. R. Vollger, J. Monlong, E. Garrison, E. E. Eichler, S. Salama, D. Haussler, R. E. Green, M. Akeson, A. Phillippy, K. H. Miga, P. Carnevali, M. Jain, and B. Paten. 2020. “Nanopore sequencing and the Shasta toolkit enable efficient de novo assembly of eleven human genomes.” Nature Biotechnology 38 (9): 1044–1053. https://doi.org/10.1038/s41587-020-0503-6.
Shu, B., and P. Gong. 2016. “Structural basis of viral RNA-dependent RNA polymerase catalysis and translocation.” Proceedings of the National Academy of Sciences 113 (28): E4005–14. https://doi.org/10.1073/pnas.1602591113.
Shu, B., and P. Gong. 2016. “Structural basis of viral RNA-dependent RNA polymerase catalysis and translocation.” Proceedings of the National Academy of Sciences 113 (28): E4005–E4014. https://doi.org/doi:10.1073/pnas.1602591113.
Shuken, S. R. 2023. “An introduction to mass spectrometry-based proteomics.” Journal of Proteome Research 22 (7): 2151–2171. https://doi.org/10.1021/acs.jproteome.2c00838.
Si-Hung, L., T. J. Causon, and S. Hann. 2017. “Comparison of fully wettable RPLC stationary phases for LC-MS-based cellular metabolomics.” Electrophoresis 38 (18): 2287–2295. https://doi.org/10.1002/elps.201700157.
Simpson, J. T., R. E. Workman, P. C. Zuzarte, M. David, L. J. Dursi, and W. Timp. 2017. “Detecting DNA cytosine methylation using nanopore sequencing.” Nature Methods 14 (4): 407–410. https://doi.org/10.1038/nmeth.4184.
Spitale, R. C., R. A. Flynn, Q. C. Zhang, P. Crisalli, B. Lee, J. W. Jung, H. Y. Kuchelmeister, P. J. Batista, E. A. Torre, E. T. Kool, and H. Y. Chang. 2015. “Structural imprints in vivo decode RNA regulatory mechanisms.” Nature 519 (7544): 486–490. https://doi.org/10.1038/nature14263.
Spitale, R. C., and D. Incarnato. 2023. “Probing the dynamic RNA structurome and its functions.” Nature Reviews Genetics 24 (3): 178–196. https://doi.org/10.1038/s41576-022-00546-w.
Stanley, J., and S. Vassilenko. 1978. “A different approach to RNA sequencing.” Nature 274 (5666): 87–89. https://doi.org/10.1038/274087a0.
Stephenson, W., R. Razaghi, S. Busan, K. M. Weeks, W. Timp, and P. Smibert. 2022. “Direct detection of RNA modifications and structure using single-molecule nanopore sequencing.” Cell Genomics 2 (2): 100097. https://doi.org/10.1016/j.xgen.2022.100097.
Stoddart, D., A. J. Heron, J. Klingelhoefer, E. Mikhailova, G. Maglia, and H. Bayley. 2010. “Nucleobase recognition in ssDNA at the central constriction of the α-hemolysin pore.” Nano Letters 10 (9): 3633–3637. https://doi.org/10.1021/nl101955a.
Stoddart, D., G. Maglia, E. Mikhailova, A. J. Heron, and H. Bayley. 2010. “Multiple base-recognition sites in a biological nanopore: Two heads are better than one.” Angewandte Chemie International Edition 49 (3): 556–559. https://doi.org/10.1002/anie.200905483.
Stoiber, M., J. Quick, R. Egan, J. Lee, S. Celniker, R. Neely, N. Loman, L. Pennacchio, and J. Brown. 2017. “De novo identification of DNA modifications enabled by genome-guided nanopore signal processing.” bioRxiv: 094672. https://doi.org/10.1101/094672
Strobel, E. J., A. M. Yu, and J. B. Lucks. 2018. “High-throughput determination of RNA structures.” Nature Reviews Genetics 19 (10): 615-634. https://doi.org/10.1038/s41576-018-0034-x.
Suzuki, T., and T. Suzuki. 2014. “A complete landscape of post-transcriptional modifications in mammalian mitochondrial tRNAs.” Nucleic Acids Research 42 (11): 7346–7357. https://doi.org/10.1093/nar/gku390.
Taucher, M., and K. Breuker. 2010. “Top-down mass spectrometry for sequencing of larger (up to 61 nt) RNA by CAD and EDD.” Journal of the American Society for Mass Spectrometry 21 (6): 918–929. https://doi.org/10.1016/j.jasms.2010.02.025.
Taucher, M., and K. Breuker. 2012. “Characterization of modified RNA by top-down mass spectrometry.” Angewandte Chemie International Edition 51 (45): 11289–11292. https://doi.org/10.1002/anie.201206232.
Tavakoli, S., M. Nabizadeh, A. Makhamreh, H. Gamper, C. A. McCormick, N. K. Rezapour, Y. M. Hou, M. Wanunu, and S. H. Rouhanifard. 2023. “Semi-quantitative detection of pseudouridine modifications and type I/II hypermodifications in human mRNAs using direct long-read sequencing.” Nature Communications 14 (1): 334. https://doi.org/10.1038/s41467-023-35858-w.
Tegowski, M., M. N. Flamand, and K. D. Meyer. 2022. “scDART-seq reveals distinct m6A signatures and mRNA methylation heterogeneity in single cells.” Molecular Cell 82 (4): 868–878.E10. https://doi.org/10.1016/j.molcel.2021.12.038.
Thakur, P., M. Estevez, P. A. Lobue, P. A. Limbach, and B. Addepalli. 2020. “Improved RNA modification mapping of cellular non-coding RNAs using C- and U-specific RNases.” Analyst 145 (3): 816–827. https://doi.org/10.1039/c9an02111f.
Thüring, K., K. Schmid, P. Keller, and M. Helm. 2016. “Analysis of RNA modifications by liquid chromatography-tandem mass spectrometry.” Methods 107: 48–56. https://doi.org/10.1016/j.ymeth.2016.03.019.
Townshend, R. J. L., S. Eismann, A. M. Watkins, R. Rangan, M. Karelina, R. Das, and R. O. Dror. 2021. “Geometric deep learning of RNA structure.” Science 373 (6558): 1047–1051. https://doi.org/doi:10.1126/science.abe5650.
Vaser, R., I. Sović, N. Nagarajan, and M. Šikić. 2017. “Fast and accurate de novo genome assembly from long uncorrected reads.” Genome Research 27 (5): 737–746. https://doi.org/10.1101/gr.214270.116.
Vilfan, I. D., Y.-C. Tsai, T. A. Clark, J. Wegener, Q. Dai, C. Yi, T. Pan, S. W. Turner, and J. Korlach. 2013. “Analysis of RNA base modification and structural rearrangement by single-molecule real-time detection of reverse transcription.” Journal of Nanobiotechnology 11 (1): 8. https://doi.org/10.1186/1477-3155-11-8.
Wagner, T. M., V. Nair, R. Guymon, S. C. Pomerantz, P. F. Crain, D. R. Davis, and J. A. McCloskey. 2004. “A novel method for sequence placement of modified nucleotides in mixtures of transfer RNA.” Nucleic Acids Symposium Series (48): 263–264. https://doi.org/10.1093/nass/48.1.263.
Wang, J., B. L. A. Chew, Y. Lai, H. Dong, L. Xu, Y. Liu, X. Y. Fu, Z. Lin, P. Y. Shi, T. K. Lu, D. Luo, S. R. Jaffrey, and P. C. Dedon. 2023. “A systems-level mass spectrometry-based technique for accurate and sensitive quantification of the RNA cap epitranscriptome.” Nature Protocols 18 (9): 2671–2698. https://doi.org/10.1038/s41596-023-00857-0.
Wang, Y., Y. Xiao, S. Dong, Q. Yu, and G. Jia. 2020. “Antibody-free enzyme-assisted chemical approach for detection of N6-methyladenosine.” Nature Chemical Biology 16 (8): 896–903. https://doi.org/10.1038/s41589-020-0525-x.
Wang, Y., S. Zhang, W. Jia, P. Fan, L. Wang, X. Li, J. Chen, Z. Cao, X. Du, Y. Liu, K. Wang, C. Hu, J. Zhang, J. Hu, P. Zhang, H. Y. Chen, and S. Huang. 2022. “Identification of nucleoside monophosphates and their epigenetic modifications using an engineered nanopore.” Nature Nanotechnology 17 (9): 976–983. https://doi.org/10.1038/s41565-022-01169-2.
Wang, Y., Y. Zhao, A. Bollas, Y. Wang, and K. F. Au. 2021. “Nanopore sequencing technology, bioinformatics and applications.” Nature Biotechnology 39 (11): 1348–1365. https://doi.org/10.1038/s41587-021-01108-x.
Watson, M., M. Thomson, J. Risse, R. Talbot, J. Santoyo-Lopez, K. Gharbi, and M. Blaxter. 2014. “poRe: An R package for the visualization and analysis of nanopore sequencing data.” Bioinformatics 31 (1): 114–115. https://doi.org/10.1093/bioinformatics/btu590.
Wein, S., B. Andrews, T. Sachsenberg, H. Santos-Rosa, O. Kohlbacher, T. Kouzarides, B. A. Garcia, and H. Weisser. 2020. “A computational platform for high-throughput analysis of RNA sequences and modifications by mass spectrometry.” Nature Communications 11 (1): 926. https://doi.org/10.1038/s41467-020-14665-7.
Wick, R. R., L. M. Judd, C. L. Gorrie, and K. E. Holt. 2017. “Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads.” PLOS Computational Biology 13 (6): E1005595. https://doi.org/10.1371/journal.pcbi.1005595.
Wick, R. R., L. M. Judd, and K. E. Holt. 2019. “Performance of neural network basecalling tools for Oxford Nanopore sequencing.” Genome Biology 20 (1): 129. https://doi.org/10.1186/s13059-019-1727-y.
Wiener, D., and S. Schwartz. 2021. “The epitranscriptome beyond m6A.” Nature Reviews Genetics 22 (2): 119–131. https://doi.org/10.1038/s41576-020-00295-8.
Wilson, C., P. J. Chen, Z. Miao, and D. R. Liu. 2020. “Programmable m6A modification of cellular RNAs with a Cas13directed methyltransferase.” Nature Biotechnology 38 (12): 1431–1440. https://doi.org/10.1038/s41587-020-0572-6.
Wolf, E. J., S. Grünberg, N. Dai, T. H. Chen, B. Roy, E. Yigit, and I. R. Corrêa. 2022. “Human RNase 4 improves mRNA sequence characterization by LC-MS/MS.” Nucleic Acids Research 50 (18): E106. https://doi.org/10.1093/nar/gkac632.
Workman, R. E., A. D. Tang, P. S. Tang, M. Jain, J. R. Tyson, R. Razaghi, P. C. Zuzarte, T. Gilpatrick, A. Payne, J. Quick, N. Sadowski, N. Holmes, J. G. de Jesus, K. L. Jones, C. M. Soulette, T. P. Snutch, N. Loman, B. Paten, M. Loose, J. T. Simpson, H. E. Olsen, A. N. Brooks, M. Akeson, and W. Timp. 2019. “Nanopore native RNA sequencing of a human poly(A) transcriptome.” Nature Methods 16 (12): 1297–1305. https://doi.org/10.1038/s41592-019-0617-2.
Wu, J., and P. Gong. 2018. “Visualizing the nucleotide addition cycle of viral RNA-dependent RNA polymerase.” Viruses 10 (1): 24. https://doi.org/10.3390/v10010024.
Xiao, Y. L., S. Liu, R. Ge, Y. Wu, C. He, M. Chen, and W. Tang. 2023. “Transcriptome-wide profiling and quantification of N6-methyladenosine by enzyme-assisted adenosine deamination.” Nature Biotechnology 41 (7): 993–1003. https://doi.org/10.1038/s41587-022-01587-6.
Xie, Y., K. A. Janssen, A. Scacchetti, E. G. Porter, Z. Lin, R. Bonasio, and B. A. Garcia. 2022. “Permethylation of ribonucleosides provides enhanced mass spectrometry quantification of post-transcriptional RNA modifications.” Analytic Chemistry 94 (20): 7246–7254. https://doi.org/10.1021/acs.analchem.2c00471.
Xue, C., Y. Zhao, and L. Li. 2020. “Advances in RNA cytosine-5 methylation: Detection, regulatory mechanisms, biological functions and links to cancer.” Biomark Research 8: 43. https://doi.org/10.1186/s40364-020-00225-0.
Xue, L., H. Yamazaki, R. Ren, M. Wanunu, A. P. Ivanov, and J. B. Edel. 2020. “Solid-state nanopore sensors.” Nature Reviews Materials 5 (12): 931–951. https://doi.org/10.1038/s41578-020-0229-6.
Yang, W., and L. Wang. 2021. “Fast and accurate algorithms for mapping and aligning long reads.” Journal of Computational Biology 28 (8): 789–803. https://doi.org/10.1089/cmb.2020.0603.
Yin, W., C. Mao, X. Luan, D. D. Shen, Q. Shen, H. Su, X. Wang, F. Zhou, W. Zhao, M. Gao, S. Chang, Y. C. Xie, G. Tian, H. W. Jiang, S. C. Tao, J. Shen, Y. Jiang, H. Jiang, Y. Xu, S. Zhang, Y. Zhang, and H. E. Xu. 2020. “Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir.” Science 368 (6498): 1499–1504. https://doi.org/10.1126/science.abc1560.
Yoluç, Y., G. Ammann, P. Barraud, M. Jora, P. A. Limbach, Y. Motorin, V. Marchand, C. Tisné, K. Borland, and S. Kellner. 2021. “Instrumental analysis of RNA modifications.” Critical Reviews in Biochemistry and Molecular Biology 56 (2): 178–204. https://doi.org/10.1080/10409238.2021.1887807.
Yu, N., M. Jora, B. Solivio, P. Thakur, C. G. Acevedo-Rocha, L. Randau, V. de Crécy-Lagard, B. Addepalli, and P. A. Limbach. 2019. “tRNA modification profiles and codon-decoding strategies in Methanocaldococcus jannaschii.” Journal of Bacteriology 201 (9). https://doi.org/10.1128/jb.00690-18.
Yu, N., P. A. Lobue, X. Cao, and P. A. Limbach. 2017. “RNAModMapper: RNA modification mapping software for analysis of liquid chromatography tandem mass spectrometry data.” Analytic Chemistry 89 (20): 10744–10752. https://doi.org/10.1021/acs.analchem.7b01780.
Zhang, Y., L. Lu, and X. Li. 2022. “Detection technologies for RNA modifications.” Experimental & Molecular Medicine 54 (10): 1601–1616. https://doi.org/10.1038/s12276-022-00821-0.
Zhang, Z., L. Q. Chen, Y. L. Zhao, C. G. Yang, I. A. Roundtree, Z. Zhang, J. Ren, W. Xie, C. He, and G. Z. Luo. 2019. “Single-base mapping of m6A by an antibody-independent method.” Science Advances 5 (7). https://doi.org/10.1126/sciadv.aax0250.
Zhang, Y. Z., A. Akdemir, G. Tremmel, S. Imoto, S. Miyano, T. Shibuya, and R. Yamaguchi. 2020. “Nanopore basecalling from a perspective of instance segmentation.” BMC Bioinformatics 21: 136. https://doi.org/10.1186/s12859-020-3459-0.
Zhang, Z., T. Chen, H. X. Chen, Y. Y. Xie, L. Q. Chen, Y. L. Zhao, B. D. Liu, L. Jin, W. Zhang, C. Liu, D. Z. Ma, G. S. Chai, Y. Zhang, W. S. Zhao, W. H. Ng, J. Chen, G. Jia, J. Yang, and G. Z. Luo. 2021. “Systematic calibration of epitranscriptomic maps using a synthetic modification-free RNA library.” Nature Methods 18 (10): 1213–1222. https://doi.org/10.1038/s41592-021-01280-7.
Zhang, M., Z. Jiang, Y. Ma, W. Liu, Y. Zhuang, B. Lu, K. Li, J. Peng, and C. Yi. 2023. “Quantitative profiling of pseudouridylation landscape in the human transcriptome.” Nature Chemical Biology 19 (10): 1185–1195. https://doi.org/10.1038/s41589-023-01304-7.
Zhao, X., Y. Zhang, D. Hang, J. Meng, and Z. Wei. 2022. “Detecting RNA modification using direct RNA sequencing: A systematic review.” Computational and Structural Biotechnology Journal 20: 5740–5749. https://doi.org/10.1016/j.csbj.2022.10.023.
Zheng, G., Y. Qin, W. C. Clark, Q. Dai, C. Yi, C. He, A. M. Lambowitz, and T. Pan. 2015. “Efficient and quantitative high-throughput tRNA sequencing.” Nature Methods 12 (9): 835–837. https://doi.org/10.1038/nmeth.3478.
Zhong, Z.-D., Y.-Y. Xie, H.-X. Chen, Y.-L. Lan, X.-H. Liu, J.-Y. Ji, F. Wu, L. Jin, J. Chen, D. W. Mak, Z. Zhang, and G.-Z. Luo. 2023. “Systematic comparison of tools used for m6A mapping from nanopore direct RNA sequencing.” Nature Communications 14 (1): 1906. https://doi.org/10.1038/s41467-023-37596-5.
Zhu, Y., S. P. Pirnie, and G. G. Carmichael. 2017. “High-throughput and site-specific identification of 2’-O-methylation sites using ribose oxidation sequencing (RibOxi-seq).” RNA 23 (8): 1303–1314. https://doi.org/10.1261/rna.061549.117.
Zimin, A. V., G. Marçais, D. Puiu, M. Roberts, S. L. Salzberg, and J. A. Yorke. 2013. “The MaSuRCA genome assembler.” Bioinformatics 29 (21): 2669–2677. https://doi.org/10.1093/bioinformatics/btt476.
Zorkot, M., R. Golestanian, and D. J. Bonthuis. 2016. “The power spectrum of ionic nanopore currents: The role of ion correlations.” Nano Letters 16 (4): 2205–2212. https://doi.org/10.1021/acs.nanolett.5b04372.
This page intentionally left blank.













































