Charting a Future for Sequencing RNA and Its Modifications: A New Era for Biology and Medicine (2024)
Chapter: Appendix F: Ideation Challenge Commissioned Papers
Appendix F
Ideation Challenge Commissioned Papers1
On June 12–13, 2023, the National Academies of Sciences, Engineering, and Medicine hosted an “Ideation Challenge” event to generate innovative approaches to solve pressing challenges in epitranscriptomics. Thirty participants from the RNA biology community formed interdisciplinary teams, identified a challenge, and discussed solving these challenges through guided sessions of collaborative brainstorming, problem solving, and providing feedback between groups. Each team recorded a video explaining their concept; these are available to view on the event website.2 A two-page written description of each concept was shared with the study committee and three teams were asked to submit white papers (see below) to describe their concepts in further detail. The commissioned papers produced by the teams guided the study committee in various aspects of the report writing process.
THE IMPACT OF RNA MODIFICATIONS ON CELLULAR FUNCTION VIA ALTERATIONS TO STRUCTURE AND MACROMOLECULAR INTERACTIONS
Author List3
Ulf Andersson Vang Ørom, Aarhus University
Nikos Tapinos, Brown University
Matthew G. Blango, Leibniz Institute for Natural Product Research and Infection Biology: Hans Knöll Institute
Jennifer Strasburger, National Human Genome Research Institute
Mark Adams, The Jackson Laboratory
___________________
1The authors are solely responsible for the content of this paper, which does not necessarily represent the views of the National Academies of Sciences, Engineering, and Medicine.
2See https://www.nationalacademies.org/event/06-12-2023/toward-sequencing-and-mapping-of-rna-modifications-ideation-challenge#sl-three-columns-ac57cecc-6691-4a03-8c4a-8dc03526113b.
3All authors contributed equally to this work.
Challenge
RNA structure and RNA modifications are aspects central to RNA function. They have both been widely recognized for decades to be important for regulatory RNAs such as transfer RNAs (tRNAs) and ribosomal RNA (rRNA), where the structure of RNA is relatively well characterized. When looking at messenger RNA (mRNA) and noncoding RNA, a much less complete picture emerges, where RNA structure is often neglected and the importance of RNA modifications is just starting to emerge (Assmann, Chou, and Bevilacqua, 2023; Zhang et al., 2022). Basic RNA structure is often depicted as a linear strand of nucleotides, sometimes with a hairpin to illustrate some degree of structure. A recent perspective article (Vicens and Kieft, 2022) very accurately highlighted the need for progression of the RNA community, as well as for the entire scientific community, towards appreciating the complexity of RNA tertiary structure; the three-dimensional arrangement of RNA building blocks that includes helical duplexes and triple-stranded structures; and the importance of structure for the molecular functions of RNA, including mRNA.
In this paper, we challenge the community to take on an ambitious effort to understand RNA structure on a transcriptome-wide level to gain new insights into the dynamics of RNA and its interaction with other macromolecules (e.g., DNA, RNA, and proteins [Figure F-1]). We emphasize the need to develop more high-throughput tools for identification of the RNA modifications currently known to occur on mRNAs in order to obtain nucleotide-precision and cell-state–dependent mapping of each modification of the transcriptome. Tools such as chemical and enzymatic conversion of specific RNA modifications and stronger direct RNA sequencing analysis pipelines are needed to meet this goal. In addition, a coordinated effort by the community to align data standards and data processing to a uniform format will be essential for driving this ambitious effort forward. RNA modifications and their impact on mRNA structure can then be assessed for their ability to mediate RNA–protein interactions (as well as interactions with RNA and DNA) with experimental and computational approaches. We encourage the community to incorporate these data into a global model that can predict RNA structure and mRNA–protein interactions and assess how these are affected by differential RNA modifications. We anticipate an “AlphaFold for RNA,” which will enlighten research into functional RNA elements, the importance of RNA modifications on basic mRNA function, and the dynamics of mRNA structure and function within the cell.

SOURCE: Created with Biorender.com.
State of the Art
The diversity of RNA modifications is demonstrated by reported functions for a diverse subset of the roughly 170 known modifications (Zhang et al., 2023), including N6-methyladenosine (m6A), N6, 2′-O-dimethyladenosine (m6Am), 5-methylcytosine (m5C), 5-hydroxymethylcytosine, inosine (I), pseudouridine (Ψ), N1-methyladenosine (m1A), 2′-O-methylation (Nm), N4-acetylcytidine (ac4C), N7-methylguanosine (m7G), dihydrouridine (D), and many others (Zhang, Lu, and Li, 2022). Different chemical modifications play distinct regulatory roles. The m6A modification influences RNA stability, splicing, translation, localization, and RNA secondary structure (Barbieri and Kouzarides, 2020; Delaunay and Frye, 2019; Shi, Wei, and He, 2019; Yang et al., 2018). Other modifications such as m5C play a role in regulating stability and translation of mRNA, while m5C regulates fidelity of translation and stability of tRNA structure (Blanco et al., 2014; Chen et al., 2019; Shanmugam et al., 2015; Shen et al., 2021; Yang et al., 2017; Yang et al., 2019; Zhang, Lu, and Li, 2022). Inosine is mainly found in double-stranded RNA (dsRNA) and plays a role in codon recoding, microRNA biogenesis, and alternative splicing site selection (Gerber and Keller, 1999; Seeburg and Hartner, 2003; Wulff, Sakurai, and Nishikura, 2011). Other modifications such as pseudouridine (Ψ) play a role in folding of rRNA, stabilize tRNA structure, and regulate biogenesis of small nuclear ribonucleoprotein (snRNP) (Arnez and Steitz, 1994; Davis, 1995; Decatur and Fournier, 2002; Jack et al., 2011; Karijolich, Yi and Yu, 2015; Karikó et al., 2008; King et al., 2003; Liang, Liu, and Fournier, 2007; Newby and Greenbaum, 2002). Introducing Ψ into mRNA results in increased protein production and alterations in the rate of translation (Eyler et al., 2019; Karijolich and Yu, 2011; Karikó et al., 2008). m1A is critical for stabilizing the tertiary structure of tRNA, while in mRNA it affects translation (Dominissini et al., 2016; Helm et al., 1998; Li, 2017; Saikia et al., 2010; Schevitz et al.,1979; Voigts-Hoffman et al., 2007). Nm plays a critical role in protein synthesis, while internal m7G has a dual role in regulating translation efficiency and enhancing micro RNA biogenesis (Choi et al., 2018; Elliot et al., 2019; Hoernes et al., 2016, 2019; Pandolfini et al., 2019; Zhang et al., 2019). Finally, ac4C induces translation of mRNA and affects biogenesis of rRNA (Arango et al., 2018; Dominissini and Rechavi, 2018; Ito et al., 2014; Jin et al., 2020; Kumbhar, Kamble, and Sonawane, 2013).
Additional features of RNA modifications have been observed for mRNA. In addition to the presence or absence of an RNA modification, the percentage of a particular transcript that is modified plays a significant role on the functional consequences of the RNA modification. For example, a modification that leads to mRNA decay is unlikely to have a significant biological effect if it affects only a small percentage of the transcriptome. Since modifications can affect mRNA structure and/or the recruitment of RNA-binding proteins, modifications of a fraction of the transcripts at any specific site would generate two distinct mRNA species that differ in their structures and protein-binding partners. Hence, changing the stoichiometry of modifications may offer another approach to generate functional diversity of mRNA.
While current RNA modification profiling methods can map the modification locations, they do not quantify the relative fraction of modified and unmodified RNA for a given transcript or phase modifications along a single RNA strand. High-throughput methods for site-specific quantification of all RNA modifications and determination of the stoichiometry of modifications will considerably advance our understanding of the functional roles of RNA modifications.
Existing Technologies—Deficiencies
Quantification of global levels of RNA modifications. Several methods for quantifying RNA modifications are already in wide use. These include dot blot assays, two-dimensional thin-layer chromatography (2D-TLC), high-performance liquid chromatography (HPLC), and liquid
chromatography–mass spectrometry (LC-MS)–based approaches, among others (Zhang, Lu, and Li, 2022). These approaches can be used to quantify the modification abundance in specific RNA species. However, they require significant amounts of RNA, which limits their utility for rare or difficult-to-acquire samples. This is especially true in the case of 2D-TLC and LC-MS, which require dedicated and expensive equipment not readily available in most laboratories around the world.
Methods for obtaining positional information of RNA modifications. Various methods have been developed for determining and quantifying the precise position of RNA modifications (Zhang, Lu, and Li, 2022).
- Primer extension. The principle of this technology is that a reverse transcriptase (RT) only reaches the 5’-end of RNA to produce a full-length complementary DNA (cDNA) if certain RNA modifications are not encountered. In the presence of modifications such as m1A, Ψ, and N1-methylguanosine, RT extension is blocked upstream of the modified site. The resulting cDNA products can then be separated by polyacrylamide gel and the terminal position of the truncated cDNA determines the position of the RNA modification. Although very sensitive, this approach has major limitations. Unfortunately, prior knowledge of the type of modification and sequence of the target RNA are required. Also, the technology can only be used for modifications that block reverse transcription and cannot be applied to “silent” modifications like m6A or m5C.
- qPCR-based approach. Semiquantitative PCR or quantitative (q)PCR–based approaches depend on the property of modified nucleotides to impede the extension of reverse transcriptase. A number of methods have been established based on these technologies to successfully detect modifications such as Nm, Ψ, N6-isopentenyladenosine, and m6A in diverse RNA species (Aschenbrenner and Marx, 2016; Castellanos-Rubio et al., 2019; Dong et al., 2012; Harcourt et al., 2013; Khalique, Mattijssen, and Maraia, 2022; Wang et al., 2021; Xiao et al., 2018; Zhang, Lu, and Li, 2022). The use of qPCR approaches has been accelerated by the development of RT enzymes that facilitate modified nucleotide detection. As one example, an engineered variant of the thermostable KlenTaq DNA polymerase harbors reverse-transcription activity and can also discriminate Nm at normal concentrations of deoxynucleoside triphosphate (Aschenbrenner and Marx, 2016; Zhang, Lu, and Li, 2022). The DNA polymerases Tth and Bst also exhibit reverse transcriptase activity and can be leveraged for locus-specific detection of m6A based on their distinct extension capabilities when encountering m6A versus A residues (Castellanos-Rubio et al., 2019; Harcourt et al., 2013; Wang et al., 2016; Zhang, Lu, and Li, 2022).
- RNase H approach. In this method, RNase H cleaves RNA into two halves at the 5′ end of the nucleotide of interest (Zhang, Lu, and Li, 2022). To achieve specificity of cleavage, a 2′-O-methyl RNA–DNA chimeric oligonucleotide is annealed to RNA. Following purification of the 3′-half of the RNA, the 5′ terminus is labeled with phosphorus-32 (32P). Finally, thin-layer chromatography is used to resolve single nucleotides after complete digestion of the 32P labeled oligonucleotides. The major limitations of this method are that it requires advanced knowledge of the sequence of interest, hazardous radioactive reagents, and specialized analytical instruments and procedures.
- Electrospray Ionization-Mass Spectrometry (ESI-MS) approach. In this method, endoribonucleases such as RNase A, RNase T1, and RNase U2 are used to digest RNA into 5–15 nucleotide fragments, which are then separated by HPLC into sequence ladders. ESI-MS is then applied to reconstruct the sequence and identify RNA modifications. The major limitation of this method is the cost of the ESI-MS and the specific expertise required for running this equipment. Another limitation is the requirement of large quantities of RNA, which makes this technique not suitable for rare RNA species or precious samples.
Next-generation sequencing methods. Several next-generation sequencing (NGS) methods have been developed for mapping RNA transcriptome-wide modifications. The methods can be divided into chemical-assisted sequencing technologies, antibody-based (immunoprecipitation) technologies, enzyme- or protein-assisted technologies, and direct sequencing technologies. Examples of these technologies validated by published methods are described below.
- Chemical-assisted sequencing. Multiple new methods are under development that can discriminate modified nucleotides from unmodified nucleotides (Zhang, Lu, and Li, 2022). So far, these approaches typically rely on the use of (1) biotin tags to enrich modified transcripts prior to sequencing; (2) altered features of base-pairing to induce misincorporation or truncation during the reverse-transcription reaction; or (3) chemical-induced cleavage, which is followed by specific adaptor ligation. More recently, an approach termed GLORI was developed that uses glyoxal and nitrite-mediated deamination of unmethylated adenosines and enables accurate and quantitative determination of m6A sites (Liu et al., 2023).
- Antibody-based technologies. This method uses antibodies specific for modifications such as m6A or m7G to immunoprecipitate modified RNA, followed by fractionation and NGS (Zhang, Lu, and Li, 2022).
- Enzyme- or protein-assisted sequencing. DART-seq uses the cytidine deaminase APOBEC1 fused to the m6A-binding YTH domain to deaminate nucleotides next to m6A-modified nucleotides (Meyer, 2019). This technology has also been used at single-cell resolution. The E. coli endoribonuclease MazF cleaves an unmethylated 5′-ACA-3′motif, but not the 5′-m6ACA-3′ motif, when followed by NGS. This method can map the presence of m6A at single-nucleotide resolution (MAZTER-seq and m6A-REF-seq) within a subset of m6A sites. The related evolved TadA-assisted N6-methyladenosine sequencing (eTAM-seq) approach relies on an optimized bacterial deaminase specific for unmodified A bases and was shown to enable m6A profiling, including from very limited RNA input amounts (Xiao et al., 2023).
- Direct RNA sequencing. Several approaches have been described using nanopore direct RNA sequencing technology coupled with artificial intelligence (AI)–based data analysis and modification identification (Cozzuto et al., 2023; Mulroney et al., 2023; Pratanwanich et al., 2021; Stephenson et al., 2022). Nanopore sequencing has also been used to detect pseudouridine semiquantitatively (Tavakoli et al., 2023). This analysis was facilitated by construction of hybrid synthetic RNA templates with base modifications at specific locations. An engineered nanopore structure with a reactive site was shown to be able to discriminate among 14 different nucleotide monophosphates containing known RNA modifications, showing that highly accurate detection of modified bases is feasible, although this system is not yet configured for sequencing of RNA strands (Wang et al., 2022).
The NGS approaches have expanded over the last couple of years at the bulk mRNA level. Major limitations are the absence of efficient and widely applicable methods for single-cell modification sequencing, as well as the limited expansion of NGS technology on modifications of other RNA species (e.g., rRNA, tRNA, circular RNA). Finally, the AI algorithms for nanopore technology and the resolution and noise deconvolution methods for using nanopore for modification-sequence mapping still need refinement, as they can only be applied for a few of the known modifications.
Short-Term Goals
There are important technological components that must be included in any plan for studying RNA modifications. These include additional investments in sequencing methods using a diversity
of approaches, the availability of standard materials for benchmarking, standard operating procedures, and appropriately organized repositories for data. However, additional near-term, foundational tasks must also be undertaken to progress to long-term goals. These tasks are as follows:
- Create affordable, reliable benchmarking materials that are widely available and coupled to accepted community standards for data analysis downstream.
- Develop easy, affordable, and accessible techniques for quantifying modifications such as m6A and the other known mRNA modifications.
- Mine microbial systems for novel enzyme potential.
- Develop libraries of enzymes for modification-specific reactions: a reverse transcriptase for each modification, endonucleases (toxin/antitoxin) to facilitate single-molecule labeling, and/or RNA-targeting CRISPR/Cas systems.
- Explore methodologies for structural assessment of modified RNAs (e.g., cross-linking and immunoprecipitation [CLIP]-seq and selective 2’-hydroxyl acylation analyzed by primer extension and variant profiling [SHAPE-MaP] approaches for modified RNAs).
Proposed methods for addressing our short-term goals:
- Quantification of global levels of RNA modifications. To address our goal of creating fast, reliable detection of RNA modifications, we need to develop easy, highly quantitative, reproducible, affordable, and expandable methods. Our proposal would be to start with methods for globally quantifying a single, abundant modification such as m6A on mRNA, and then expand to other modifications as technology and community standards mature. For an easy, quantitative, and affordable method of quantifying m6A on mRNA, we believe that efforts should focus on qPCR-based approaches (e.g., immuno-PCR), since these are easy to use, highly quantitative, require minimal starting material (so they can be used with rare or difficult to obtain samples), and can be adopted by laboratories around the world. Independent of the approach that is ultimately successful for this purpose, we will need clear and robust experimental and data standards that are agreed upon and adopted by the user community.
- Positional information and NGS approaches. To obtain the tools for specifically mapping and sequencing all RNA modifications, we propose several tasks. (1) We see large potential in mining bacterial systems and/or enzymes used by bacteria as defense mechanisms against other pathogens to identify RNA-targeting enzymes that recognize and modify (or protect) specific RNA motifs harboring individual modifications, such as those implemented in eTAM-seq. Another example comes from the bacterial (E. coli, Mycobacterium tuberculosis) toxin-antitoxin systems, which include the endoribonuclease MazF, which is capable of recognizing m6A-modified motifs on mRNA (Zhang et al., 2003, 2005; Zhu et al., 2006, 2008) and also cleaving rRNA (Schifano and Woychik, 2014; Schifano et al., 2013, 2014) and certain tRNAs (Schifano et al., 2016). Similar approaches have been used on a global RNA-seq level with techniques such as MORE-seq, which determines target sites of MazF (Schifano et al., 2024). (2) Recently, two approaches to map and quantify single-base m6A in the mammalian transcriptome have been introduced. GLORI-seq (Liu et al., 2023) and eTAM-seq (Xiao et al., 2023) use enzyme-assisted adenosine deamination to discriminate methylated from unmethylated adenosines at a single-nucleotide resolution. These techniques, although they avoid the false positive signals obtained by MazF and the limited MazF cleavage to ACA motifs, cannot discriminate between m6Am and m1A, are more expensive than traditional MeRIP-seq, and are limited to m6A. Development of similar enzyme-assisted approaches that discriminate between other modified and
- unmodified nucleotides at single-base resolution will significantly enhance our understanding of the biological importance and role of RNA modifications in mammalian systems. Finally, (3) we need to perform a systematic search for reverse transcriptases or DNA polymerases with RT activity (e.g., KlenTag and Marathon RT [de Cesaris Araujo Tavares et al., 2023]) to discover enzymes that can be engineered to detect individual RNA modifications; and (4) pursue discovery of RNA-specific CRISPR-Cas systems capable of recognizing and preferentially editing or cleaving modified versus unmodified RNA. The recent discovery of the Class VI CRISPR-Cas system effector, Cas13, shown to target RNA (Abudayyeh et al., 2020; East-Seletsky et al., 2016; Shmakov et al., 2015) makes this approach a promising option.
- Single-cell NGS for RNA modifications. We propose employing the enzyme-assisted sequencing described above (e.g., MAZTER-seq), by using newly discovered enzymes from bacterial systems to cleave single-cell RNA on certain modifications depending on the recognition specificity of the enzyme. The fragmented RNA could then be barcoded, or adaptor ligated, and sequenced at a single-cell level. Since this is still a laborious technology with a significant limitation for the number of cells that can be sequenced, we propose to develop recombinant enzymes as fusion enzymes with endoribonuclease activity and tagmentation capabilities (e.g., MazF-Tn5). These enzymes should be able to generate tagmented, modified RNA fragments that could be pooled and sequenced based on limitless combination of indexes and primers used for tagmentation, a technology reminiscent of combinatorial indexing.
Achieving these goals will require coordinated efforts from many laboratories and disciplines, using multiple complementary or orthogonal methods and approaches pursued in parallel to address the range of modifications in mRNA and their effects on structure and interactions. While we propose initial, proof-of-principle studies focused on mRNA and well-characterized modifications such as m6A, the methods we establish here should be expandable into other subsets of RNA—tRNA, rRNA, long noncoding RNA—as well as to other RNA modifications.
Long-Term Goals
The challenge of defining the impact of RNA modifications on RNA cellular function is a big task and will likely advance our understanding of RNA-modification biology in multiple directions. These will be punctuated by a series of long-term goals over the next 5–10 years:
- Develop single-cell (or low-abundance) modification sequencing to study structure at the single-cell level.
- Determine impact of RNA modifications on interactions of RNA with proteins (including readers, writers, and erasers of RNA modifications), other RNAs, and DNA sequences.
- Predict RNA structure based on modifications and sequence, including through use of molecular dynamic simulations and machine learning, and experimental approaches that probe RNA structure.
- Assess impact of structure on function.
Proposed methods for addressing long-term goals.
- Develop single-cell (or low abundance) modification sequencing to study base modifications at the single-cell level.
Existing technologies. The advent of single-cell RNA-seq (scRNA-seq) technology has brought about a lowered detection limit for rare RNA species, and facilitated new, previously unimaginable investigations. In conjunction with additional molecular techniques, scRNA-seq can now be coupled to chromatin accessibility, surface protein expression, and even spatial information (Heumos et al., 2023; Longo et al., 2021; Williams et al., 2022). Techniques such as the recently released RiboMap have even allowed translation to be monitored in a large subset of genes at spatial resolution across individual cells (Zeng et al., 2023). These applications of scRNA-seq are representative of the potential of sequencing individual RNAs in a single cell and provide encouragement for ultimately reaching the level of the single-cell or even single-molecule epitranscriptome. In fact, a recent example goes so far as to measure m6A levels in vivo, in single large cells (e.g., embryonic stem cells, zebrafish zygotes, mouse oocytes), suggesting that, in principle, our goal is not so far from reality (Li et al., 2023). The recent emergence of third-generation sequencing technologies such as Oxford Nanopore’s direct RNA sequencing is groundbreaking, but we still have room to grow. Early work has facilitated sequencing of individual RNAs, albeit with relatively high starting concentrations of RNA; has expanded our understanding of RNA processing; and has even allowed detection of some modifications, including m6A (Abebe et al., 2022; Jain et al., 2022; Stephenson et al., 2022). Unfortunately, error rates remain relatively high, secondary structure of the RNA can be a challenge, and the sensitivity is still lacking in most use cases (Leger et al., 2021; Liu, Begik, and Novoa, 2021; Liu-Wei et al., 2023; Zhang, Lu, and Li, 2022); however, the recent release of new, direct RNA-seq chemistry by Oxford Nanopore has started to address some of these issues. Bioinformatic strategies for processing scRNA-seq and direct RNA sequencing have greatly improved, but work remains to understand RNA modifications of a single molecule within a single cell (Cozzuto et al., 2020).
Specific actions. The first long-term goal we propose is to advance technology to be able to sequence modifications in individual cells and/or from low concentrations of RNA input. The added value for such an advance is obvious, as increased sensitivity of scRNA-seq will certainly increase our ability to detect RNA modifications, particularly rare or novel modifications that may only be observable in disease states. To achieve this goal, more reliable library preparation methods will likely be required, in addition to novel bioinformatic approaches to “deconvoluting” the data. We can imagine applications in conjunction with rapidly emerging AI technologies, which we suspect will greatly impact our day-to-day routines in the 5- to 10-year timespan. We also anticipate that increases in sensitivity and reliability of Oxford Nanopore direct RNA sequencing, and other platforms, will be required to reach this long-term goal. A major push in the field is already underway, but refined efforts towards defining best practices and providing benchmarking materials will be essential in providing a robust solution. For this purpose, better spike-in controls for modified RNAs will be needed to optimize approaches and more accurately identify RNA modifications in single cells and/or low-abundance samples.
Reliance on short-term goals. The improvement of scRNA-seq and direct RNA-seq to detect RNA modifications in low-abundance samples such as single cells will be built on the foundation established in our short-term goals. It will be essential to develop easy, affordable, and accessible techniques for quantifying modifications, such as m6A as a first use case. We appreciate that the solution to reliable, robust single-cell, single-molecule modification sequencing may require completely novel technology. We expect many advances in this direction to continue to come from mining of microbial systems, which have been repeatedly shown to carry vast potential. As work on understanding microbial communities continues, we expect that novel enzyme activities will be identified, for example, from nonmodel organisms, particularly the understudied fungi. The development of libraries of enzymes for modification-specific reactions will also likely facilitate new approaches, as has occurred with the examples discussed above.
- Determine impact of RNA modifications on interactions of RNA with proteins (including readers, writers, and erasers of RNA modifications), other RNAs, and DNA sequences.
Existing technologies. RNA modifications have been studied for decades to great success. Unfortunately, we still have an insufficient understanding of the role of RNA modifications in the interactions between modified RNA and other macromolecules. Certainly, examples exist, but the tools to understand these interactions on a large scale remain relatively absent. For example, we know that editing by proteins like adenosine deaminase acting on RNA (ADAR) can result in inosine-containing RNAs with altered innate immune activities (Liao et al., 2011; Sarvestani et al., 2014). Modified tRNAs were shown to fit more snuggly into the ribosome, suggesting alterations to interactions between the modified tRNA and ribosomal proteins/RNA (Dannfald, Favory, and Deragon, 2021; Hoffer et al., 2020; Yasukawa et al., 2001), and even mRNA methylation marks influence binding of downstream factors, including m6A core machinery and additional peripheral factors (Lewis, Pan, and Kolsotra, 2017; Shi, Wei, and He, 2019). These examples hint at regulation of biological function, but we also know that RNA modifications can influence RNA structure. Again, the best examples likely come from studing tRNA modifications, where installed modifications have been shown on numerous occasions to stabilize tRNA tertiary structure (Biedenbänder et al., 2022). RNA modifications are also thought to influence RNA:DNA hybrids after damage and during R-loop formation, hinting that a systemic description of the rules of interaction of the epitranscriptome with all cellular macromolecules holds vast potential in advancing our molecular understanding of the influence of RNA modifications on cellular interactions and function (Abakir et al., 2020).
Specific actions. To systematically define the influence of RNA modifications on intermolecular interactions, the field will need to strive for several key advances. First, a robust set of biochemical assays must be established and thoroughly tested to facilitate measurement of interactions between individual modified RNAs and other macromolecules. These initial biochemical assays should be extended to broad-scale analysis, likely requiring efforts to improve large-scale synthesis of modified RNAs for massively parallel reporter assays. Additional efforts in high-throughput synthesis of modifications at many sites simultaneously followed by structure elucidation will also be essential. It will be important to develop targeted RNA editing platforms that can be tunable to further introduce marks and determine structures.
Reliance on short-term goals. To succeed with the second long-term goal, we will need to develop easy, affordable, and accessible techniques for quantifying modifications as described above. The short-term goals described are necessary advances to characterize robustly the importance of modifications in intermolecular interactions. Again, as above, we see potential in mining microbial systems for novel enzyme potential, and we suspect that additional novel solutions will be found within the vast microbial pangenome. Many other tasks must also be completed, but we see value in developing tools that will further facilitate this long-term goal—for example, by developing libraries of enzymes for modification-specific reactions, endonucleases to facilitate single-molecule labeling, and RNA-targeting CRISPR/Cas systems that could be used for reading out modified RNAs or even intermolecular interactions.
- Predict RNA structure based on modifications and sequence, including through use of molecular dynamic simulations and machine learning.
Existing technologies. In recent years, cryo-electron microscopy technology and structural probing methods have greatly improved our understanding of tertiary interactions in RNA structure (Theil, Flamm, and Hofacker, 2017), yet in many cases RNA prediction remains the logical first step in understanding potential function. As such, a wide array of RNA prediction tools have been
developed to address this particular challenge (Mfold, ViennaFold/RNAfold, and many others) (Piechotta et al., 2022; Tsybulskyi, Semenchenko, and Meyer, 2023). These tools tend to rely on thermodynamic models, such as Turner’s nearest-neighbor model to predict secondary structures by focusing on nearest-neighbor loops (Sato, Akiyama, and Sakakibara, 2021). These can then be combined to find the minimum-free energy structure using the Zuker algorithm (Zuker, 1989). As we continue to obtain more experimentally validated structures, these predictions are more frequently based on reference molecules, which has improved prediction (Do, Woods, and Batzoglou, 2006; Zakov et al., 2011). The inclusion of advanced computational approaches is also pushing our prediction capabilities forward, for example by leveraging deep learning and neural networks (Booy, Ilin, and Orponen, 2022; Chen and Chan, 2023; Sato, Akiyama, and Sakakibara, 2021; Singh et al., 2019; Townshend et al., 2021). For the most part, the role of modifications in influencing tertiary, or even secondary, structure, has been difficult to incorporate into these predictions, but some efforts have been made (reviewed in Tanzer, Hofacker, and Lorenz, 2019), including prediction of structures with well-characterized modifications like m6A (Kierzek et al., 2022).
Specific actions. To successfully predict RNA tertiary structure for RNAs containing modifications, a much larger reference set of experimentally validated RNA structures is needed. As with AlphaFold for proteins, the use of AI can allow for valid, informative predictions with enough input, but our current cohort of RNA structures remains far too limited to saturate the complexity of RNA structures, especially RNA molecules containing modified nucleotides. In achieving this goal, we will continue to need organized, curated databases of high-quality structures to efficiently integrate large amounts of data into better learning predictions. In fact, some amazing examples of RNA structure and dynamics have already been predicted (Yu et al., 2021), but these computationally heavy predictions already push our computing power to the limit, suggesting additional advances are necessary. With recent successes in quantum computing, we expect that this technology, or even another currently unappreciated approach, will lead to sufficient resources for better predictions in the future. Although beyond the scope of this paper, performing such energy intensive computational activities should be developed with climate neutral strategies in mind.
Reliance on short-term goals. The recent progress in approaches like CLIP-seq (Hafner et al., 2021) and SHAPE-MaP (Bohn et al., 2023; Smola and Weeks, 2018) have facilitated a more rapid assessment of RNA structure in the laboratory, but additional approaches to more rapidly assess these structures in conjunction with RNA modifications are certainly needed. Our short-term goal in exploring additional methodologies for structural assessment of modified RNAs will be essential to improving predictions. Clearly, a better toolbox of benchmarking materials will also facilitate the structural determination of modified RNAs, allowing for the field to move closer to a state where an AlphaFold-like solution will be able to robustly, rapidly predict all RNA molecules.
- Assess impact of structure on function (as controlled by modifications).
Existing technologies. The fourth long-term goal strives to provide a resolution for the overall goal of understanding the functional consequences of RNA editing on RNA function, particularly during disease. RNA modifications are already known to control normal cellular homeostatic processes (Frye et al., 2018), contribute to regulation or dysregulation of cellular processes as during cancers (Haruehanroengra et al., 2020), and even influence the outcome of infection, both by regulating host immune processes and pathogen virulence (Cui et al., 2022). Modifications of tRNAs control translational fidelity (Agris et al., 2017), editing marks installed by ADAR and APOBEC3 proteins control autoimmunity and antiviral activity (Liu et al., 2018; Quin et al., 2021), and ribosomal RNAs are severely dysfunctional in the absence of modifications resulting in devastating human diseases (reviewed in Sharma and Entian, 2022). If we consider the m6A RNA modification as one example, we already a substantial amount (Sendinc and Shi, 2023). This prolific mark has
clear roles in the function of mRNAs as well as noncoding RNAs, but also contributes to regulation of housekeeping RNAs like rRNA. These functions collectively contribute to regulation of chromatin architecture, transcriptional regulation, and genome stability (Boulias and Greer, 2023). In fact, m6A methylation can also directly influence protein binding, for example by preventing binding of proteins to particular sites. A clear example of this regulation comes from C. elegans, where environmental growth conditions trigger the METT-10 writer to modify the 3’ splice site of S-adenosylmethionine synthetase to prevent binding of the essential splicing factor U2AF35 and splicing (Mendel et al., 2021). Many other examples show similar regulation of RNA modifications across biology, yet despite this clear importance, in most situations, RNA editing is barely considered in regulation.
Specific actions. Should our dream come to fruition, we expect to have the tools to produce a detailed description of RNA modifications on a single molecule of RNA in a single cell, which would facilitate the elucidation of cellular consequences of such modifications. Clearly a tall order, smaller steps again must be taken to reach this comprehensive understanding. We believe that striving to define a single, simple system more fully (e.g., a cell type selected from Encode Tier 1) will ultimately facilitate our larger goals. By perturbing such a system (e.g., with nutrient deprivation, genetic manipulation, temperature change, infection), we expect to understand the dynamics of modifications and the cellular consequences of RNA modifications. We expect that advances along the way will push analyses like these in many directions, beyond single-cell types to tissues or even whole organisms. By establishing workflows and techniques for a complete description the impact of RNA modifications on structure to control function in one cell type, we expect to facilitate easier investigations more broadly.
Reliance on short-term goals. Clearly the final long-term goal will rely on input from many of the other pieces of our approach. The success of the short-term goals, especially the creation of easy, affordable, and accessible techniques for quantifying modifications such as N6-methyladenosine (m6A) will be critical in reaching a fully described cellular example of RNA modification impact on function. At the moment, it is not clear how to achieve such a goal, but we expect that advancements in enzymatic studies, including novel activities mined from microbes, will facilitate our success.
Future Technologies
Several technologies already exist that can be leveraged for the success of this project. These include nuclear magnetic resonance, cryo-electron microscopy, X-Ray Crystallography, multiple sequencing strategies (including mass spectrometry, nanopore sequencing, and affinity capture methods), and a variety of bioinformatics approaches (e.g., machine learning) as described above. In order to realize these goals, a strategy must be established to incorporate existing technologies more robustly, support collaborations, promote the scientific exchange of ideas and foster development of new technologies. It is clear that the technology does not exist to fully address our stated challenge, but we can at least partially envision the advances required to reach these goals. We will certainly need alternative enzyme activities, which we can envisage being revealed through exploration of the microbial world or synthetic efforts to evolve or design new functionalities in existing enzymatic scaffolds. Among these activities, we anticipate the need for enzymes capable of barcoding modified RNA. Many laboratories and companies are already exploring this space, and we are cautiously optimistic that we are on the cusp of a revolution in RNA enzymology. Although we expect new functionalities and entirely new solutions, we do expect advances to continue from current workhorses, particularly with the rapid advancement in technologies surrounding the use of CRISPR-Cas-like approaches to specifically add/remove modifications. Synthetic biology tools and programmable RNA editing technologies may help establish or generate standardized reagents (reviewed in Booth et al., 2023; Xu et al., 2022).
We believe that the strategic, specific, and reliable installation of RNA modifications on synthetic RNA molecules is an advancing technology, but it still needs additional effort to realize a robust method to create useful standards for the types of experiments we are proposing. Synthesis of long RNAs with multiple modifications introduced at specified positions at proper stoichiometry would be a big advantage but remains nearly impossible. There are some promising leads in this direction, for example with techniques relying on enzymatic splint ligation (Gramper et al., 2022). With all these gains in the wet lab, we will also need to keep pace with our computational infrastructure.
If Moore’s law continues to hold true, we expect to see advances in computing power, as well as cleverly designed databases to support sharing of information on RNA modifications, structures, and interactions with appropriate metadata. Many discussions are currently underway, but key among them will be resources to maintain databases over long stretches of time, especially as computational techniques and supercomputing capabilities increase the amount of data collected from each experiment. Finally, we expect that optimization of experimental designs may also result in improvements in data output. As just one example, new approaches are constantly being explored to increase depth of coverage, as sequencing the same sample more deeply does not always reveal new information (e.g., modification sites or unique RNA molecules). We therefore expect that additional approaches will be developed to extract information from the datasets we have in hand, as well as to improve collection in new experiments.
Societal Relevance
Stakeholders
Depending upon where any biomedical research falls on the spectrum of basic to clinical, stakeholders in a research endeavor can include collaborators, peer reviewers, research institutions, funding agencies, clinicians and professional associations, patients, and policy makers (Concannon et al., 2019; Cottrell et al., 2014; Garrison et al., 2021). Collaborators can be actively engaged in a research endeavor, provide an important resource, or act in an advisory capacity. Peer reviewers, whether their purview is study protocols, manuscripts, or applications for funding, are engaged as stakeholders in upholding research rigor and ethical conduct. Research institutions, whether for-profit or not-for-profit entities, are a fundamental driver of research innovation and development. These institutions are often early incubators of patentable technologies developed from research. Research institutions, particularly those with graduate programs and professional schools, invest heavily in equipment, facilities, and personnel in support of the research activities of their faculty (Rosowsky, 2022). Biomedical and clinical research, especially those studies involving clinical trials, can be expensive. Much of this research is funded by federal agencies such as the National Science Foundation, the National Institutes of Health, the U.S. Department of Energy, and Department of Defense, but organizations such as foundations and philanthropies support smaller research studies, too. Patients are increasingly seen as stakeholders in research involving their illnesses and medical care. Patient input provides important information for the development of essential aspects of a clinical study such as a study’s design and outcome measures. The importance of patient stakeholder engagement has been recognized and is oftentimes mandated or strongly endorsed by research funders and journals (Harrison et al., 2019). Policy makers include government agencies at the federal, state, and local levels as well as other funders of research. Policies based upon the interpretation of outcomes of major research endeavors can have a significant impact on public health (for example, vaccine development for COVID-19). Policy makers also determine research priorities and funding levels.
Ultimately, the more heavily invested—intellectually or financially—a collaborator, an institution, a funder, etc. is in a research project, the greater the stake that individual or group may feel they have in the success of that project.
Beneficiaries
In terms of potential economic impact, the beneficiaries of biomedical research would be widely ranging, from basic researchers, to patients, to local and national governments. In the short term, other basic researchers would benefit the most quickly from improved technologies in RNA structure prediction, molecular interaction detection, and direct RNA sequencing. For other groups pursuing clinical applications, the benefit will come in the long term after requisite steps, such as developing strategies to promote or block RNA modification-directed processes, clinical trials, and new drug development. In all cases, near-term and long-term benefit is dependent on the dissemination and adoption of any novel, cutting-edge technologies that are developed (Dearing and Cox, 2018).
Workforce, Educational, and Societal Considerations
There is disagreement within the scientific community on the “benefit” of research and development. Some express strong views that progress in science and technology, when enabled by regressive science policy practices, may make life harder for people, especially those already marginalized by class, gender, race, occupation, and location. One example is that scientific innovation, such as new therapeutics, mainly benefits those that can afford it (Bozeman, 2020). Others argue that public investment in science translates to public use of science, which is seen as a “benefit.” One such study analyzed the scientific literature and identified positive correlations between funding in a research area and scientific use. This extended to public use as well (Yin, 2022). At minimum, stronger public engagement in research planning and in the discussion of research outcomes may increase public perception of the importance and positive impact of RNA research on human health and medicine.
Workforce development, interdisciplinary training, and diversity and inclusion are certainly areas of critical need discussed at multiple, recent scientific venues, including a May 2022 workshop organized by the NIH, NHGRI, and NIEHS, entitled “Capturing RNA Sequence and Transcript Diversity, From Technology Innovation to Clinical Application” (NIEHS, 2022). The following were among several relevant points captured in the executive summary and report for the workshop.
- Support of both undergraduate and graduate-level training, especially at minority-serving institutions, could help improve diversity within the biomedical research community.
- Training in both “wet-lab” experimental methods and “dry-lab” computational biology and bioinformatics is critical for the advancement and widespread adoption of RNA technologies.
- Easily accessible, quality, low (or no)-cost, online training resources on technology use and data analysis could be a considerable driver in technology adoption.
- Enhancing career pathways for all groups, including those underrepresented in biomedical research, is important. Considerations include identifying current successful career enhancement approaches that could be scaled up, and determining resources and strategies needed to address ongoing barriers to advancement.
Anticipated Outcome
The end goal of the project is to understand how RNA modifications affect RNA function, including processing and interactions with other macromolecules on a transcriptome-wide scale. We can measure how completely we have addressed this challenge by assessing the RNA community’s ability to establish standards and common analysis pipelines to allow for a high degree of collaboration. This can also be supported by the level of usage of public data and sharing of data between groups. To accomplish this goal, we need an even higher degree of open data policies and data sharing between laboratories working in all aspects of the biology of RNA modifications.
We can assess the progress of single-cell technologies directly and in parallel with the depth and accuracy available for mapping and quantification of the various RNA modifications. A more challenging aspect to assess is the prediction of RNA structure and the influence of RNA modifications on structure and interactions with macromolecules. While the field is expected to continuously progress, when can we really claim that we have reached our goal? Certainly, we will have come a long way when tertiary structure can be predicted from primary sequence and the impact of a subset of the predominant mRNA modifications are faithfully incorporated in predictions (which are subsequently experimentally validated). Adding the ability to predict dynamic interactions with proteins accurately and reliably on a large scale, especially proteins involved in RNA processing, will bring us across the finish line.
The four long-term goals proposed will have tremendous impact on the RNA community, if accomplished, and they will provide unprecedented insight into the dynamics of RNA function and biological importance of RNA modifications. The development of single-cell knowledge of RNA modification identity and stoichiometry will reveal novel regulatory functions for subsets of RNA transcripts previously thought to be identical based on sequence information. With additional RNA modification information, we are likely to appreciate new flexible and dynamic conformations in these RNAs, in part mediated by differential RNA modification, which can mediate a multitude of previously unappreciated functions.
The ability to predict tertiary structure of RNA based on sequence and modification pattern will be a break-through for RNA research and will help establish RNA as an attractive molecule for several other disciplines to study. The ability to faithfully predict dynamic and context-dependent protein interactions will reveal new details of molecular biology with interest for RNA biologists, biochemists, protein researchers, and cell biologists, whereas an approach to predict RNA interaction with small molecules on a high level would revolutionize the potential of RNA medicine and provide new candidate targets for a plethora of diseases.
Conclusion
The Human Genome Project revealed the blueprint for building a human; however, the DNA sequence is just the beginning. We now must strive to understand the role of RNA in this construction process and fully elucidate the function of RNA in the cell, in all its modalities. The modifications introduced during RNA metabolism are considered by many to be a whole new level of gene expression regulation. By deciphering the regulatory code of RNA modifications, we will more completely understand the full potential of this macromolecule, unlock new functionalities for RNA, and likely provide novel therapeutic ingress points to the tremendous benefit of humanity. It is an undertaking worthy of continued research effort and investment.
REFERENCES
Abakir, A., T. C. Giles, A. Cristini, , J. M. Foster, N. Dai, M. Starczak, A. Rubio-Roldan, M. Li, M. Eleftheriou, J. Crutchley, L. Flatt, L. Young, D. J. Gaffney, C. Denning, B. Dalhus, R. D. Emes, D. Gackowski, I. R. Corrêa, J. L. Garcia-Perez Jr., A. Klungland, N. Gromak, and A. Ruzov. 2020. “N6-methyladenosine regulates the stability of RNA:DNA hybrids in human cells.” Nature Genetics 52(1), 48–55. https://doi.org/10.1038/s41588-019-0549-x.
Abebe, J. S., A. M. Price, K.E. Hayer, I. Mohr, M.D. Weitzman, A. C. Wilson, and D. P. Depledge. 2022. “DRUMMER-rapid detection of RNA modifications through comparative nanopore sequencing.” Bioinformatics 38(11), 3113–3115. https://doi.org/10.1093/bioinformatics/btac274.
Abudayyeh, O. O., J. S. Gootenberg, S. Konermann, J. Joung, I. M. Slaymaker, D. B. Cox, S. Shmakov, K. S. Makarova, E. Semenova, L. Minakhin, K. Severinov, A. Regev, E. S. Lander, E. V. Koonin, and F. Zeng. 2016. “C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector.” Science 353(6299):AAF5573. https://doi.org/10.1126/science.aaf5573.
Agris, P. F., A. Narendran, K. Sarachan, V. Y. P. Väre, and E. Eruysal. 2017. “The importance of being modified: The role of RNA modifications in translational fidelity.” The Enzymes 41:1–50. https://doi.org/10.1016/bs.enz.2017.03.005.
Arango, D., D. Sturgill, N. Alhusaini, A. A. Dillman, T. J. Sweet, G. Hanson, M. Hosogane, W. R. Sinclair, K. K. Nanan, M. D. Mandler, S. D. Fox, T. T. Zengeya, T. Andresson, J. L. Meier, J. Coller, and S. Oberdoerffer. 2018. “Acetylation of Cytidine in mRNA promotes translation efficiency.” Cell 175(7):1872–1886.E24. https://doi.org/10.1016/j.cell.2018.10.030.
Arnez, J. G., and T. A. Steitz. 1994. “Crystal structure of unmodified tRNA(Gln) complexed with glutaminyl-tRNA synthetase and ATP suggests a possible role for pseudo-uridines in stabilization of RNA structure.” Biochemistry 33(24):7560–7567. https://doi.org/10.1021/bi00190a008.
Aschenbrenner, J., and A. Marx. 2016. “Direct and site-specific quantification of RNA 2’-O-methylation by PCR with an engineered DNA polymerase.” Nucleic Acids Research 44(8):3495–3502. https://doi.org/10.1093/nar/gkw200.
Assmann, S. M., H-L. Chou, and P. C. Bevilacqua. 2023. “Rock, scissors, paper: How RNA structure informs function.” The Plant Cell 35(6):1671–1707. https://doi.org/10.1093/plcell/koad026.
Barbieri, I., and T. Kouzarides. 2020. “Role of RNA modifications in cancer.” Nature Reviews Cancer 20(6):303–322. https://doi.org/10.1038/s41568-020-0253-2.
Biedenbänder, T., V. de Jesus, M. Schmidt-Dengler, M. Helm, B. Corzilius, and B. Fürtig. 2022. “RNA modifications stabilize the tertiary structure of tRNAfMet by locally increasing conformational dynamics.” Nucleic Acids Research 50(4): 2334–2349. https://doi.org/10.1093/nar/gkac040.
Blanco, S., S. Dietmann, J. V. Flores, S. Hussain, C. Kutter, P. Humphreys, M. Lukk, P. Lombard, L. Treps, M. Popis, S. Kellner, S. M. Hölter, L. Garrett, W. Wurst, L. Becker, T. Klopstock, H. Fuchs, V. Gailus-Durner, M. Hrabĕ de Angelis, R. T. Káradóttir, M. Helm, J. Ule, J. G. Gleeson, D. T. Odom, and M. Frye. 2014. “Aberrant methylation of tRNAs links cellular stress to neurodevelopmental disorders.” EMBO Journal 33(18):2020–2039. https://doi.org/10.15252/embj.201489282
Bohn, P., A. S. Gribling-Burrer, U. B. Ambi, and R. P. Smyth. 2023. “Nano-DMS-MaP allows isoform-specific RNA structure determination.” Nature Methods 20(6):849–859. https://doi.org/10.1038/s41592-023-01862-7.
Booth, B. J., S. Nourreddine, D. Katrekar, Y. Savva, D. Bose, T. J. Long, D. J. Huss, and P. Mali. 2023. “RNA editing: Expanding the potential of RNA therapeutics.” Molecular Therapy 31(6): 1533–1549. https://doi.org/10.1016/j.ymthe.2023.01.005.
Booy, M. S., A. Ilin, and P. Orponen. 2022. “RNA secondary structure prediction with convolutional neural networks.” BMC Bioinformatics 23(1):58. https://doi.org/10.1186/s12859-021-04540-7.
Boulias, K., and E. L. Greer. 2023. “Biological roles of adenine methylation in RNA.” Nature Reviews Genetics 24(3):143–160. https://doi.org/10.1038/s41576-022-00534-0.
Bozeman, B. 2020. “Public Value Science.” Issues in Science and Technology 36(4) https://issues.org/public-value-science-innovation-equity-bozeman/ (accessed July 28, 2023).
Castellanos-Rubio, A., I. Santin, A. Olazagoitia-Garmendia, I. Romero-Garmendia, A. Jauregi-Miguel, M. Legarda, and J. R. Bilbao. 2019. “A novel RT-QPCR-based assay for the relative quantification of residue specific m6A RNA methylation.” Scientific Reports 9(1):4220. https://doi.org/10.1038/s41598-019-40018-6.
Chen, C-C., and Y-M. Chan. 2023. “REDfold: Accurate RNA secondary structure prediction using residual encoder-decoder network.” BMC Bioinformatics 24(1):122. https://doi.org/10.1186/s12859-023-05238-8.
Chen, X., A. Li, B-F. Sun, Y. Y. Yang, Y-N. Han, X. Yuan, R-X. Chen, W-S. Wei, Y. Liu, C-C. Gao, Y-S. Chen, M. Zhang, X-D. Ma, Z-W. Liu, J-H. Luo, C. Lyu, H-L. Wang, J. Ma, Y-L. Zhao, F-J. Zhou, Y. Huang, D. Xie, and Y-G. Yang. 2019. “5-methylcytosine promotes pathogenesis of bladder cancer through stabilizing mRNAs.” Nature Cell Biology 21(8):978–990. https://doi.org/10.1038/s41556-019-0361-y.
Choi, J., G. Indrisiunaite, H. DeMirci, K-W. Ieong, J. Wang, A. Petrov, A. Prabhakar, G. Rechavi, D. Dominissini, C. He, M. Ehrenberg, and J. D. Puglisi. 2018. “2′-O-methylation in mRNA disrupts tRNA decoding during translation elongation.” Nature Structural & Molecular Biology 25(3):208–216. https://doi.org/10.1038/s41594-018-0030-z.
Concannon, T. W., S. Grant, V. Welch, J. Petkovic, J. Selby, S. Crowe, A. Synnot, R. Greer-Smith, E. Mayo-Wilson, E. Tambor, P. Tugwell, and Multi Stakeholder Engagement (MuSE) Consortium. 2019. “Practical guidance for involving stakeholders in health research.” Journal of General Internal Medicine 34(3):458–463. https://doi.org/10.1007/s11606-018-4738-6.
Cottrell, E., E. Whitlock, E. Kato, S. Uhl, S. Belinson, C. Chang, T. Hoomans, D. Meltzer, H. Noorani, K. Robinson, K. Schoelles, M. Motu’apuaka, J. Anderson, R. Paynter, and J-M. Guise. 2014. Defining the benefits of stakeholder engagement in systematic reviews. Rockville, MD: Agency for Healthcare Research and Quality. http://www.ncbi.nlm.nih.gov/books/NBK196180/.
Cozzuto, L., A. Delgado-Tejedor, T. Hermoso Pulido, E. M. Novoa, and J. Ponomarenko. 2023. “Nanopore direct RNA sequencing data processing and analysis using MasterOfPores.” Methods in Molecular Biology 2624:185–205. https://doi.org/10.1007/978-1-0716-2962-8_13.
Cozzuto, L., H. Liu, L. P. Pryszcz, T. H. Pulido, A. Delgado-Tejedor, J. Ponomarenko, and E. M. Novoa. 2020. “MasterOfPores: A workflow for the analysis of Oxford Nanopore direct RNA sequencing datasets.” Frontiers in Genetics 11:211. https://doi.org/10.3389/fgene.2020.00211.
Cui, L., R. Ma, J. Cai, C. Guo, Z. Chen, L. Yao, Y. Wang, R. Fan, X. Wang, and Y. Shi. 2022. “RNA modifications: Importance in immune cell biology and related diseases.” Signal Transduction and Targeted Therapy 7(1):334. https://doi.org/10.1038/s41392-022-01175-9.
Dannfald, A., J-J. Favory, and J-M. Deragon. 2021. “Variations in transfer and ribosomal RNA epitranscriptomic status can adapt eukaryote translation to changing physiological and environmental conditions.” RNA Biology 18(Suppl 1):4–18. https://doi.org/10.1080/15476286.2021.1931756.
Davis, D.R. 1995. “Stabilization of RNA stacking by pseudouridine.” Nucleic Acids Research 23(24):5020–5026. https://doi.org/10.1093/nar/23.24.5020.
de Cesaris Araujo Tavares, R., G. Mahadeshwar, H. Wan, and A. M. Pyle. 2023. “MRT-ModSeq– Rapid detection of RNA modifications with MarathonRT.” Journal of Molecular Biology https://doi.org/10.1101/2023.05.25.542276 (accessed July 31, 2023).
Dearing, J. W., and J. G. Cox. 2018. “Diffusion of innovations theory, principles, and practice.” Health Affairs 37(2):183–190. https://doi.org/10.1377/hlthaff.2017.1104.
Decatur, W. A., and M. J. Fournier. 2002. “rRNA modifications and ribosome function.” Trends in Biochemical Sciences 27(7):344–351. https://doi.org/10.1016/s0968-0004(02)02109-6.
Delaunay, S., and M. Frye. 2019. “RNA modifications regulating cell fate in cancer.” Nature Cell Biology 21(5):552–559. https://doi.org/10.1038/s41556-019-0319-0.
Do, C. B., D. A. Woods, and S. Batzoglou. 2006. “CONTRAfold: RNA secondary structure prediction without physics-based models.” Bioinformatics 22(14):E90-98. https://doi.org/10.1093/bioinformatics/btl246.
Dominissini, D., S. Nachtergaele, S. Moshitch-Moshkovitz, E. Peer, N. Kol, M. S. Ben-Haim, Q. Dai, A. Di Segni, M. Salmon-Divon, W. C. Clark, G. Zheng, T. Pan, O. Solomon, E. Eyal, V. Hershkovitz, D. Han, L. C. Doré, N. Amariglio, G. Rechavi, and C. He. 2016. “The dynamic N(1)-methyladenosine methylome in eukaryotic messenger RNA.” Nature 530(7591):441–446. https://doi.org/10.1038/nature16998.
Dominissini, D., and G. Rechavi. 2018. “N4-acetylation of cytidine in mRNA by NAT10 regulates stability and translation.” Cell 175(7):1725–1727. https://doi.org/10.1016/j.cell.2018.11.037.
Dong, Z-W., P. Shao, L-T. Diao, H. Zhou, C-H. Yu, and L-H. Qu. 2012. “RTL-P: A sensitive approach for detecting sites of 2’-O-methylation in RNA molecules.” Nucleic Acids Research 40(20):E157.
East-Seletsky, A., M. R. O’Connell, S. C. Knight, D. Burstein, J. H. Cate, R. Tjian, and J. A. Doudna. 2016. “Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection.” Nature 538(7624):270–273. https://doi.org/10.1038/nature19802.
Elliott, B. A., H-T. Ho, S. V. Ranganathan, S. Vangaveti, O. Ilkayeva, H. Abou Assi, A. K. Choi, P. F. Agris, and C. L. Holley. 2019. “Modification of messenger RNA by 2′-O-methylation regulates gene expression in vivo.” Nature Communications 10(1):3401. https://doi.org/10.1038/s41467-019-11375-7.
Eyler, D. E., M. K. Franco, Z. Batool, M. Z. Wu, M. L. Dubuke, M. Dobosz-Bartoszek, J. D. Jones, Y. S. Polikanov, B. Roy, and K. S. Koutmou. 2019. “Pseudouridinylation of mRNA coding sequences alters translation.” Proceedings of the National Academy of Sciences of the United States of America 116(46):23068–23074. https://doi.org/10.1073/pnas.1821754116.
Frye, M., B. T. Harada, M. Behm, and C. He. 2018. “RNA modifications modulate gene expression during development.” Science 361(6409):1346–1349. https://doi.org/10.1126/science.aau1646.
Gamper, H., C. McCormick, S. Tavakoli, M. Wanunu, S. H. Rouhanifard, and Y-M. Hou. 2022. “Synthesis of long RNA with a site-specific modification by enzymatic splint ligation.” https://doi.org/10.1101/2022.09.17.508400.
Garrison, H., M. Agostinho, L. Alvarez, S. Bekaert, L. Bengtsson, E. Broglio, D. Couso, R. Araújo Gomes, Z. Ingram, E. Martinez, A. Lúcia Mena, D. Nickel, M. Norman, I. Pinherio, M. Solís-Mateos, and M. G. Bertero. 2021. “Involving society in science: Reflections on meaningful and impactful stakeholder engagement in fundamental research.” EMBO Reports 22(11):E54000. https://doi.org/10.15252/embr.202154000.
Gerber, A.P., and W. Keller. 1999. “An adenosine deaminase that generates inosine at the wobble position of tRNAs.” Science 286(5442):1146–1149. https://doi.org/10.1126/science.286.5442.1146.
Hafner, M., M. Katsantoni, T. Köster, J. Marks, J. Mukherjee, D. Staiger, J. Ule, and M. Zavolan. 2021. “CLIP and complementary methods.” Nature Reviews Methods Primers 1(1):1–23. https://doi.org/10.1038/s43586-021-00018-1.
Harcourt, E. M., T. Ehrenschwender, P J. Batista, H. Y. Chang, and E. T. Kool. 2013. “Identification of a selective polymerase enables detection of N(6)-methyladenosine in RNA.” Journal of the American Chemical Society 135(51):19079–19082. https://doi.org/10.1021/ja4105792.
Harrison, J. D., A. D. Auerbach, W. Anderson, M. Fagan, M. Carnie, C. Hanson, J. Banta, G. Symczak, E. Robinson, J. Schnipper, C. Wong, and R. Weiss. 2019. “Patient stakeholder engagement in research: A narrative review to describe foundational principles and best practice activities.” Health Expectations 22(3):307–316. https://doi.org/10.1111/hex.12873.
Haruehanroengra, P., Y. Y. Zheng, Y. Zhou, Y. Huang, and J. Sheng. 2020. “RNA modifications and cancer.” RNA Biology 17(11):1560–1575. https://doi.org/10.1080/15476286.2020.1722449.
Helm, M., H. Brulé, F. Degoul, C. Cepanec, J. P. Leroux, R. Giegé, and C. Florentz. 1998. “The presence of modified nucleotides is required for cloverleaf folding of a human mitochondrial tRNA.” Nucleic Acids Research 26(7):1636–1643. https://doi.org/10.1093/nar/26.7.1636.
Heumos, L., A. C. Schaar, C. Lance, A. Litinetskaya, F. Drost, L. Zappia, M. D. Lücken, D. C. Strobl, J. Henao, F. Curion, Single-cell Best Practices Consortium, H. B. Schiller, and F. J. Theis. 2023. “Best practices for single-cell analysis across modalities.” Nature Reviews Genetics 24(8):550–572. https://doi.org/10.1038/s41576-023-00586-w.
Hoernes, T. P., N. Clementi, K. Faserl, H. Glasner, K. Breuker, H. Lindner, A. Hüttenhofer, and M. D. Erlacher. 2016. “Nucleotide modifications within bacterial messenger RNAs regulate their translation and are able to rewire the genetic code.” Nucleic Acids Research 44(2):852–862. https://doi.org/10.1093/nar/gkv1182.
Hoernes, T. P., D. Heimdörfer, D. Köstner, K. Faserl, F. Nußbaumer, R. Plangger, C. Kreutz, H. Lindner, and M. D. Erlacher. 2019. “Eukaryotic translation elongation is modulated by single natural nucleotide derivatives in the coding sequences of mRNAs.” Genes 10(2):84. https://doi.org/10.3390/genes10020084.
Hoffer, E. D., S. Hong, S. Sunita, T. Maehigashi, R. L. Gonzalez, P. C. Whitford, and C. M. Dunham. 2020. “Structural insights into mRNA reading frame regulation by tRNA modification and slippery codon-anticodon pairing.” eLife 9:E51898. https://doi.org/10.7554/eLife.51898.
Ito, S., Y. Akamatsu, A. Noma, S. Kimura, K. Miyauchi, Y. Ikeuchi, T. Suzuki, and T. Suzuki. 2014. “A single acetylation of 18 S rRNA is essential for biogenesis of the small ribosomal subunit in Saccharomyces cerevisiae.” Journal of Biological Chemistry 289(38):26201–26212. https://doi.org/10.1074/jbc.M114.593996.
Jack, K., C. Bellodi, D. M. Landry, R. O. Niederer, A. Meskauskas, S. Musalgaonkar, N. Kopmar, O. Krasnykh, A. M Dean, S. R. Thompson, D. Ruggero, and J. D. Dinman. 2011. “rRNA pseudouridylation defects affect ribosomal ligand binding and translational fidelity from yeast to human cells.” Molecular Cell 44(4):660–666. https://doi.org/10.1016/j.molcel.2011.09.017.
Jain, M., R. Abu-Shumays, H. E. Olsen, and M. Akeson. 2022. “Advances in nanopore direct RNA sequencing.” Nature Methods 19(10):1160–1164. https://doi.org/10.1038/s41592-022-01633-w.
Jin, G., M. Xu, M. Zou, and S. Duan. 2020. “The processing, gene regulation, biological functions, and clinical relevance of N4-acetylcytidine on RNA: A systematic review.” Molecular Therapy- Nucleic Acids 20:13–24. https://doi.org/10.1016/j.omtn.2020.01.037.
Karijolich, J., C. Yi, and Y-T. Yu. 2015. “Transcriptome-wide dynamics of RNA pseudouridylation.” Nature Reviews Molecular Cell Biology 16(10):581–585. https://doi.org/10.1038/nrm4040.
Karijolich, J., and Y-T. Yu. 2011. “Converting nonsense codons into sense codons by targeted pseudouridylation.” Nature 474(7351):395–398. https://doi.org/10.1038/nature10165.
Karikó, K., H. Muramatsu, F. A. Welsh, J. Ludwig, H. Kato, S. Akira, and D. Weissman. 2008. “Incorporation of Pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability.” Molecular Therapy 16(11):1833–1840. https://doi.org/10.1038/mt.2008.200.
Khalique, A., S. Mattijssen, and R. J. Maraia. 2022. “A versatile tRNA modification-sensitive northern blot method with enhanced performance.” RNA 28(3):418–432. https://doi.org/10.1261/rna.078929.121.
Kierzek, E., X. Zhang, R. M. Watson, S. D. Kennedy, M. Szabat, R. Kierzek, and D. H. Mathews. 2022. “Secondary structure prediction for RNA sequences including N6-methyladenosine.” Nature Communications 13(1): 1271. https://doi.org/10.1038/s41467-022-28817-4.
King, T. H., B. Liu, R. R. McCully, and M. J. Fournier. 2003. “Ribosome structure and activity are altered in cells lacking snoRNPs that form pseudouridines in the peptidyl transferase center.” Molecular Cell 11(2):425–435. https://doi.org/10.1016/s1097-2765(03)00040-6.
Kumbhar, B. V., A. D. Kamble, and K. D. Sonawane. 2013. “Conformational preferences of modified nucleoside N(4)acetylcytidine, ac4C occur at ‘wobble’ 34th position in the anticodon loop of tRNA.” Cell Biochemistry and Biophysics 66(3):797–816. https://doi.org/10.1007/s12013-013-9525-8.
Leger, A., P. P. Amaral, L. Pandolfini, C. Capitanchik C, F. Capraro, V. Miano, V. Migliori, P. Toolan-Kerr, T. Sideri, A. J. Enright, K. Tzelepis, F. J. van Werven, N. M. Luscombe, I. Barbieri, J. Ule, T. Fitzgerald, E. Birney, T. Leonardi, and T. Kouzarides. 2021. “RNA modifications detection by comparative Nanopore direct RNA sequencing.” Nature Communications 12(1):7198. https://doi.org/10.1038/s41467-021-27393-3.
Lewis, C.J., T. Pan, and A. Kalsotra. 2017. “RNA modifications and structures cooperate to guide RNA-protein interactions.” Nature Reviews Molecular Cell Biology 18(3):202–210. https://doi.org/10.1038/nrm.2016.163.
Li, X., X. Xiong, M. Zhang, K. Wang, Y. Chen, J. Zhou, Y. Mao, J. Lv, D. Yi, X-W. Chen, C. Wang, S-B. Qian, and C. Yi. 2017. “Base-resolution mapping reveals distinct m1A methylome in nuclear- and mitochondrial-encoded transcripts.” Molecular Cell 68(5):993-1005.E9. https://doi.org/10.1016/j.molcel.2017.10.019.
Li, Y., Y. Wang, M. Vera-Rodriguez, L. C. Lindeman, L. E. Skuggen, E. M. K. Rasmussen, I. Jermstad, S. Khan, M. Fosslie, T. Skuland, M. Indahl, S. Khodeer, E. K. Klemsdal, K-X. Jin, K. T. Dalen, P. Fedorcsak, G. D. Greggains, M. Lerdrup, A. Klugland, K. F. Au, and J. A. Dahl. 2023. “Single-cell m6A mapping in vivo using picoMeRIP-seq.” Nature Biotechnology. https://doi.org/10.1038/s41587-023-01831-7.
Liang, X-H., Q. Liu, and M. J. Fournier. 2007. “rRNA modifications in an intersubunit bridge of the ribosome strongly affect both ribosome biogenesis and activity.” Molecular Cell 28(6):965–977. https://doi.org/10.1016/j.molcel.2007.10.012
Liao, J-Y., S. A. Thakur, Z. B. Zalinger, K. E. Gerrish, and F. Imani. 2011. “Inosine-containing RNA is a novel innate immune recognition element and reduces RSV infection.” PLoS ONE 6(10):E26463. https://doi.org/10.1371/journal.pone.0026463
Liu, C., H. Sun, Y. Yi, W. Shen, K. Li, Y. Xiao, F. Li, Y. Li, Y. Hou, B. Lu, W. Liu, H. Meng, J. Peng, C. Yi, and J. Wang. 2023. “Absolute quantification of single-base m6A methylation in the mammalian transcriptome using GLORI.” Nature Biotechnology 41(3):355–366. https://doi.org/10.1038/s41587-022-01487-9
Liu, H., O. Begik, and E. M. Novoa. 2021. “EpiNano: Detection of m6A RNA modifications using Oxford Nanopore direct RNA sequencing.” Methods in Molecular Biology 2298:31–52. https://doi.org/10.1007/978-1-0716-1374-0_3.
Liu, M-C., W-Y. Liao, K. M. Buckley, S. Y. Yang, J. P. Rast, and S. D. Fugmann. 2018. “AID/APOBEC-like cytidine deaminases are ancient innate immune mediators in invertebrates.” Nature Communications 9(1):1948. https://doi.org/10.1038/s41467-018-04273-x
Liu-Wei, W., W. van der Toorn, P. Bohn, M. Hölzer, R. Smyth, and M. von Kleist. 2023. Sequencing accuracy and systematic errors of nanopore direct RNA sequencing. ttps://doi.org/10.1101/2023.03.29.534691.
Longo, S. K., M. G. Guo, A. L. Ji, and P. A. Khavari. 2021. “Integrating single-cell and spatial transcriptomics to elucidate intercellular tissue dynamics.” Nature Reviews Genetics 22(10):627–644. https://doi.org/10.1038/s41576-021-00370-8
Mendel, M., K. Delaney, R. R. Pandey, K-M. Chen, J. M. Wenda, C. B. Vågbø, F. A. Steiner, D. Homolka, and R. S. Pillai. 2021. “Splice site m6A methylation prevents binding of U2AF35 to inhibit RNA splicing.” Cell 184(12):3125-3142. E25. https://doi.org/10.1016/j.cell.2021.03.062.
Meyer, K. D. 2019. “DART-seq: An antibody-free method for global m6A detection.” Nature Methods 16(12):1275–1280. https://doi.org/10.1038/s41592-019-0570-0.
Mulroney, L., E. Birney, T. Leonardi, and F. Nicassio. 2023. “Using Nanocompore to identify RNA modifications from direct RNA Nanopore sequencing data.” Current Protocols 3(2):E683. https://doi.org/10.1002/cpz1.683.
Newby, M.I., and N. L. Greenbaum. 2002. “Investigation of Overhauser effects between pseudouridine and water protons in RNA helices.” Proceedings of the National Academy of Sciences of the United States of America 99(20):12697–12702. https://doi.org/10.1073/pnas.202477199.
NIEHS (National Institute of Environmental Health Sciences). 2022. Capturing RNA Sequence and Transcript Diversity, From Technology Innovation to Clinical Application. Virtual event occurring May 24–26, 2022. https://www.niehs.nih.gov/news/events/pastmtg/2022/rnaworkshop2022/index.cfm (accessed July 24, 2023).
Pandolfini, L., I. Barbieri, A. J. Bannister, A. Hendrick, B. Andrews, N. Webster, P. Murat, P. Mach, R. Brandi, S. C. Robson, V. Migliori, A. Alendar, M. d’Onofrio, S. Balasubramanian, and T. Kouzarides. 2019. “METTL1 promotes let-7 MicroRNA processing via m7G methylation.” Molecular Cell 74(6):1278–1290.E9. https://doi.org/10.1016/j.molcel.2019.03.040.
Piechotta, M., I. S. Naarmann-de Vries, Q. Wang, J. Altmüller, and C. Dieterich. 2022. “RNA modification mapping with JACUSA2.” Genome Biology 23(1):115. https://doi.org/10.1186/s13059-022-02676-0.
Pratanwanich, P. N., F. Yao, Y. Chen, C. W. Q. Koh, Y. K. Wan, C. Hendra, P. Poon, Y. T. Goh, P. M. L. Yap, J. Y. Chooi, W. J. Chng, S. B. Ng, A. Thiery, W. S. S. Goh, and J. Göke. 2021. “Identification of differential RNA modifications from nanopore direct RNA sequencing with xPore.” Nature Biotechnology 39(11):1394–1402. https://doi.org/10.1038/s41587-021-00949-w.
Quin, J., J. Sedmík, D. Vukić, A. Khan, L. P. Keegan, and M. A. O’Connell. 2021. “ADAR RNA modifications, the epitranscriptome and innate immunity.” Trends in Biochemical Sciences 46(9):758–771. https://doi.org/10.1016/j.tibs.2021.02.002.
Rosowsky, D. 2022. “The role of research at universities: Why it matters.” Forbes. https://www.forbes.com/sites/davidro-sowsky/2022/03/02/the-role-of-research-at-universities-why-it-matters/ (accessed July 28, 2023).
Saikia, M., Y. Fu, M. Pavon-Eternod, C. He, and T. Pan. 2010. “Genome-wide analysis of N1-methyl-adenosine modification in human tRNAs.” RNA 16(7):1317–1327. https://doi.org/10.1261/rna.2057810.
Sarvestani, S. T., M. D. Tate, J. M. Moffat, A. M. Jacobi, M. A. Behlke, A. R. Miller, S. A. Beckham, C. E. McCoy, W. Chen, J. D. Mintern, M. O’Keeffe, M. John, B. R. G. Williams, and M. P. Gantier. 2014. “Inosine-mediated modulation of RNA sensing by Toll-like receptor 7 (TLR7) and TLR8.” Journal of Virology 88(2):799–810.
Sato, K., M. Akiyama, and Y. Sakakibara. 2021. “RNA secondary structure prediction using deep learning with thermodynamic integration.” Nature Communications 12(1):941. https://doi.org/10.1038/s41467-021-21194-4.
Schevitz, R. W., A. D. Podjarny, N. Krishnamachari, J. J. Hughes, P. B. Sigler, and J. L. Sussman. 1979. “Crystal structure of a eukaryotic initiator tRNA.” Nature 278(5700):188–190. https://doi.org/10.1038/278188a0.
Schifano, J. M., J. W. Cruz, I. O. Vvedenskaya, R. Edifor, M. Ouyang, R. N. Husson. 2016. “tRNA is a new target for cleavage by a MazF toxin.” Nucleic Acids Research 44(3):1256–1270. https://doi.org/10.1093/nar/gkv1370.
Schifano, J. M., R. Edifor, J. D. Sharp, M. Ouyang, A. Konkimalla, R. N. Husson, and N. A. Woychik. 2013. “Mycobacterial toxin MazF-mt6 inhibits translation through cleavage of 23S rRNA at the ribosomal A site.” Proceedings of the National Academy of Sciences of the United States of America 110(21):8501–8506. https://doi.org/10.1073/pnas.1222031110.
Schifano, J. M., I. O. Vvedenskaya, J. G. Knoblauch, M. Ouyang, B. E. Nickels, and N. A. Woychik. 2014. “An RNA-seq method for defining endoribonuclease cleavage specificity identifies dual rRNA substrates for toxin MazF-mt3.” Nature Communications 5:3538. https://doi.org/10.1038/ncomms4538.
Schifano, J. M., and N. A. Woychik. 2014. “23S rRNA as an a-Maz-ing new bacterial toxin target.” RNA Biology 11(2):101–105. https://doi.org/10.4161/rna.27949.
Seeburg, P. H., and J. Hartner. 2003. “Regulation of ion channel/neurotransmitter receptor function by RNA editing.” Current Opinion in Neurobiology 13(3):279–283. https://doi.org/10.1016/s0959-4388(03)00062-x
Sendinc, E., and Y. Shi. 2023. “RNA m6A methylation across the transcriptome.” Molecular Cell 83(3):428–441. https://doi.org/10.1016/j.molcel.2023.01.006.
Shanmugam, R., J. Fierer, S. Kaiser, M. Helm, T. P. Jurkowski, and A. Jeltsch. 2015. “Cytosine methylation of tRNA-Asp by DNMT2 has a role in translation of proteins containing poly-Asp sequences.” Cell Discovery 1(1):1–10. https://doi.org/10.1038/celldisc.2015.10.
Sharma, S., and K-D. Entian. 2022. “Chemical modifications of ribosomal RNA.” Methods in Molecular Biology 2533:149–166. https://doi.org/10.1007/978-1-0716-2501-9_9.
Shen, H., R. J. Ontiveros, M. C. Owens, M. Y. Liu, U. Ghanty, R. M. Kohli, and K. F. Liu. 2021. “TET-mediated 5-methylcy-tosine oxidation in tRNA promotes translation.” Journal of Biological Chemistry 296:100087. https://doi.org/10.1074/jbc.ra120.014226.
Shi, H., J. Wei, and C. He. 2019. “Where, when and how: Context-dependent functions of RNA methylation writers, readers, and erasers.” Molecular Cell 74(4):640–650. https://doi.org/10.1016/j.molcel.2019.04.025.
Shmakov, S., O. O. Abudayyeh, K. S. Makarova, Y. I. Wolf, J. S. Gootenberg, E. Semenova, L. Minakhin, J. Joung, S. Konermann, K. Severinov, F. Zhang, and E. V. Koonin. 2015. “Discovery and functional characterization of diverse Class 2 CRISPR-Cas systems.” Molecular Cell 60(3):385–397. https://doi.org/10.1016/j.molcel.2015.10.008.
Singh, J., J. Hanson, K. Paliwal, and Y. Zhou. 2019. “RNA secondary structure prediction using an ensemble of two-dimensional deep neural networks and transfer learning.” Nature Communications 10(1):5407. https://doi.org/10.1038/s41467-019-13395-9.
Smola, M. J., and K. M. Weeks. 2018. “In-cell RNA structure probing with SHAPE-MaP.” Nature Protocols 13(6):1181–1195. https://doi.org/10.1038/nprot.2018.010.
Stephenson, W., R. Razaghi, S. Busan, K. M. Weeks, W. Timp, and P. Smibert. 2022. “Direct detection of RNA modifications and structure using single-molecule nanopore sequencing.” Cell Genomics 2(2):100097. https://doi.org/10.1016/j.xgen.2022.100097.
Tanzer, A., I. L. Hofacker, and R. Lorenz. 2019. “RNA modifications in structure prediction – Status quo and future challenges.” Methods 156:32–39. https://doi.org/10.1016/j.ymeth.2018.10.019.
Tavakoli, S., M. Nabizadeh, A. Makhamreh, H. Gamper, C. A. McCormick, N. K. Rezapour, Y-M. Hou, M. Wanunu, and S. H. Rouhanifard. 2023. “Semi-quantitative detection of pseudouridine modifications and type I/II hypermodifications in human mRNAs using direct long-read sequencing.” Nature Communications 14(1):334. https://doi.org/10.1038/s41467-023-35858-w.
Thiel, B. C., C. Flamm, and I. L. Hofacker. 2017. “RNA structure prediction: From 2D to 3D.” Emerging Topics in Life Sciences 1(3):275–285. https://doi.org/10.1042/ETLS20160027.
Townshend, R. J. L., S. Eismann, A. M. Watkins, R. Rangan, M. Karelina, R. Das, and R. O. Dror. 2021. “Geometric deep learning of RNA structure.” Science 373(6558):1047–1051. https://doi.org/10.1126/science.abe5650.
Tsybulskyi, V., E. Semenchenko, and I. M. Meyer. 2023. “e-RNA: A collection of web-servers for the prediction and visualisation of RNA secondary structure and their functional features.” Nucleic Acids Research 51(W1):W160–167. https://doi.org/10.1093/nar/gkad296.
Vicens, Q., and J. S. Kieft. 2022. “Thoughts on how to think (and talk) about RNA structure.” Proceedings of the National Academy of Sciences of the United States of America 119(17):E2112677119. https://doi.org/10.1073/pnas.2112677119.
Voigts-Hoffmann, F., M. Hengesbach, A. Y. Kobitski, A. van Aerschot, P. Herdewijn, G. U. Nienhaus, and M. Helm. 2007. “A methyl group controls conformational equilibrium in human mitochondrial tRNA(Lys).” Journal of the American Chemical Society 129(44):13382–13383. https://doi.org/10.1021/ja075520+.
Wang, S., J. Wang, Z. Zhang, B. Fu, Y. Song, P. Ma, K. Gu, X. Zhou, X. Zhang, T. Tian, and X. Zhou. 2016. “N6-Methyladenine hinders RNA- and DNA-directed DNA synthesis: Application in human rRNA methylation analysis of clinical specimens.” Chemical Science 7(2):1440–1446. https://doi.org/10.1039/c5sc02902c.
Wang, Y., S. Zhang, W. Jia, P. Fan, L. Wang, X. Li, J. Chen, Z. Cao, X. Du, Y. Liu, K. Wang, C. Hu, J. Zhang, J. Hu, P. Zhang, H-Y. Chen, and S. Huang. 2022. “Identification of nucleoside monophosphates and their epigenetic modifications using an engineered nanopore.” Nature Nanotechnology 17(9):976–983. https://doi.org/10.1038/s41565-022-01169-2.
Wang, Y., Z. Zhang, C. Sepich-Poore, L. Zhang, Y. Xiao, and C. He. 2021. “LEAD-m6A-seq for Locus-Specific Detection of N6-Methyladenosine and Quantification of Differential Methylation.” Angewandte Chemie 60(2):873–880.
Williams, C. G., H. J. Lee, T. Asatsuma, R. Vento-Tormo, and A. Haque. 2022. “An introduction to spatial transcriptomics for biomedical research.” Genome Medicine 14(1):68. https://doi.org/10.1186/s13073-022-01075-1.
Wulff, B-E., M. Sakurai, and K. Nishikura. 2011. “Elucidating the inosinome: Global approaches to adenosine-to-inosine RNA editing.” Nature Reviews Genetics 12(2):81–85. https://doi.org/10.1038/nrg2915.
Xiao, Y., Y. Wang, Q. Tang, L. Wei, X. Zhang, and G. Jia. 2018. “An elongation- and ligation-based qPCR amplification method for the radiolabeling-free detection of locus-specific N6 -Methyladenosine modification.” Angewandte Chemie 57(49):15995–6000. https://doi.org/10.1002/anie.201807942.
Xiao, Y-L., S. Liu, R. Ge, Y, Wu, C. He, M. Chen M, and W. Tang. 2023. “Transcriptome-wide profiling and quantification of N6-methyladenosine by enzyme-assisted adenosine deamination.” Nature Biotechnology 41(7):993–1003. https://doi.org/10.1038/s41587-022-01587-6.
Xu, W., J. Biswas, R. H. Singer, and M. Rosbash. 2022. “Targeted RNA editing: Novel tools to study post-transcriptional regulation.” Molecular Cell 82(2):389–403. https://doi.org/10.1016/j.molcel.2021.10.010.
Yang, X., Y. Yang, B-F. Sun, Y-S. Chen, J-W. Xu, W-Y. Lai, A. Li, X. Wang, D. P. Bhattarai, W. Xiao, H-Y. Sun, Q. Zhu, H-L. Ma, S. Adhikari, M. Sun, Y-J. Hao, B. Zhang, C-M. Huang, N. Huang, G-B. Jiang, Y-L. Zhao, H-L. Wang, Y-P. Sun, and Y-G. Yang. 2017. “5-methylcytosine promotes mRNA export - NSUN2 as the methyltransferase and ALYREF as an m5C reader.” Cell Research 27(5):606–625. https://doi.org/10.1038/cr.2017.55.
Yang, Y., P. J. Hsu, Y-S. Chen, and Y-G. Yang. 2018. “Dynamic transcriptomic m6A decoration: Writers, erasers, readers and functions in RNA metabolism.” Cell Research 28(6):616–624. https://doi.org/10.1038/s41422-018-0040-8.
Yang, Y., L. Wang, X. Han, W-L. Yang, M. Zhang, H-L. Ma, B-F. Sun, A. Li, J. Xia, J. Chen, J. Heng, B. Wu, Y-S. Chen, J-W. Xu, X. Yang, H. Yao, J. Sun, C. Lyu, H-L. Wang, Y. Huang, Y-P. Sun, Y-L. Zhao, A. Meng, J. Ma, F. Liu, and Y-G. Yang. 2019. “RNA 5-Methylcytosine facilitates the maternal-to-zygotic transition by preventing maternal mRNA decay. Molecular Cell 75(6):1188–1202.E11. https://doi.org/10.1016/j.molcel.2019.06.033.
Yasukawa, T., T. Suzuki, N. Ishii, S. Ohta, and K. Watanabe. 2001. “Wobble modification defect in tRNA disturbs codon-anticodon interaction in a mitochondrial disease.” The EMBO Journal 20(17):4794–4802. https://doi.org/10.1093/emboj/20.17.4794.
Yin, Y., Y. Dong, K. Wang, D. Wang, and B. F. Jones. 2022. “Public use and public funding of science.” Nature Human Behavior 6(10):1344–1350. https://doi.org/10.1038/s41562-022-01397-5.
Yu, A. M., P. M. Gasper, L. Cheng, L. B. Lai, S. Kaur, V. Gopalan, A. A. Chen, and J. B. Lucks. 2021. “Computationally reconstructing cotranscriptional RNA folding from experimental data reveals rearrangement of non-native folding intermediates.” Molecular Cell 81(4):870-883E10. https://doi.org/10.1016/j.molcel.2020.12.017.
Zakov, S., Y. Goldberg, M. Elhadad, and M. Ziv-Ukelson. 2011. “Rich parameterization improves RNA structure prediction.” Journal of Computational Biology 18(11):1525–1542. https://doi.org/10.1089/cmb.2011.0184.
Zeng, H., J. Huang, J. Ren, C. K. Wang, Z. Tang, H. Zhou, Y. Zhou, H. Shi, A. Aditham, X. Sui, H. Chen, J. A. Lo, and X. Wang. 2023. “Spatially resolved single-cell translatomics at molecular resolution.” Science 380(6652). https://doi.org/10.1126/science.add3067.
Zhang, J., Y. Fei, L. Sun, and Q. C. Zhang. 2022. “Advances and opportunities in RNA structure experimental determination and computational modeling.” Nature Methods 19(10):1193–1207. https://doi.org/10.1038/s41592-022-01623-y.
Zhang, L-S., C. Liu, H. Ma, Q. Dai, H-L. Sun, G. Luo, Z. Zhang, L. Zhang, L. Hu, Z. Dong, and C. He. 2019. “Transcrip-tome-wide mapping of internal N7-methylguanosine methylome in mammalian mRNA.” Molecular Cell 74(6):1304–1316.E8. https://doi.org/10.1016/j.molcel.2019.03.036.
Zhang, Y., L. Lu, and X. Li. 2022. “Detection technologies for RNA modifications.” Experimental & Molecular Medicine 54(10):1601–1616. https://doi.org/10.1038/s12276-022-00821-0.
Zhang, Y., J. Jiang, J. Ma, Z. Wei, Y. Wang, B. Song, J. Meng, G. Jia, J. P. de Magalhães, D. J Rigden, D. Hang, and K. Chen. 2023. “DirectRMDB: A database of post-transcriptional RNA modifications unveiled from direct RNA sequencing technology.” Nucleic Acids Research 51(D1):D106–D116. https://doi.org/10.1093/nar/gkac1061.
Zhang, Y., J. Zhang, K. P. Hoeflich, M. Ikura, G. Qing, and M. Inouye. 2003. “MazF cleaves cellular mRNAs specifically at ACA to block protein synthesis in Escherichia coli.” Molecular Cell 12(4):913–923. https://doi.org/10.1016/s1097-2765(03)00402-7.
Zhang, Y., J. Zhang, H. Hara, I. Kato, and M. Inouye. 2005. “Insights into the mRNA cleavage mechanism by MazF, an mRNA interferase.” Journal of Biological Chemistry 280(5):3143–3150. https://doi.org/10.1074/jbc.M411811200.
Zhu, L., S. Phadtare, H. Nariya, M. Ouyang, R. N. Husson, and M. Inouye. 2008. “The mRNA interferases, MazF-mt3 and MazF-mt7 from Mycobacterium tuberculosis target unique pentad sequences in single-stranded RNA.” Molecular Microbiology 69(3):559–569. https://doi.org/10.1111/j.1365-2958.2008.06284.x.
Zhu, L., Y. Zhang, J-S. The, J. Zhang, N. Connell, H. Rubin, and M. Inouye. 2006. “Characterization of mRNA interferases from Mycobacterium tuberculosis.” Journal of Biological Chemistry 281(27):18638–18643. https://doi.org/10.1074/jbc.m512693200.
Zuker, M. 1989. “On finding all suboptimal foldings of an RNA molecule.” Science 244(4900):48–52. https://doi.org/10.1126/science.2468181.
VISION FOR A UNIFIED EPITRANSCRIPTOMICS CONSORTIUM (EPIC)
Author List4
Norman H. L. Chiu, The University of North Carolina at Greensboro
Dragony Fu, University of Rochester
Siddhardha S. Maligireddy, National Cancer Institute, National Institutes of Health
Charles P. Rabolli, The Ohio State University
Nikolaos Tsotakos, The Pennsylvania State University
Jonah Z. Vilseck, Indiana University School of Medicine
Introduction
The completion of the Human Genome Project represented a monumental advancement in our understanding of the regulation and evolution of human genes. In addition to a publicly available and programmatically accessible record of the completed human genome, the new research methods and technologies developed to carry out the project transformed how other fields approached genetics. Several years later, the publication of the ModENCODE project illuminated the functional elements in eukaryotic genomes and further demonstrated that collaboration between labs and institutions is crucial for tackling large-scale questions in biology.
Due to the complexity of gene regulation, the scientific community has now devoted its efforts to understanding the next frontiers of regulatory mechanisms that lie beyond the genome. Epigenetic alterations play a significant regulatory role in gene expression, ranging from structural changes in chromosomes to the methylation of individual DNA base pairs. At the protein level, post-translational modifications to the peptide sequence of a protein significantly impact its structure and function. Finally, RNA, processed and modified through various mechanisms such as capping, splicing, editing, and chemical modifications, plays important roles in gene regulation and cellular function.
The field of biology has become increasingly interested in the chemical modification of individual RNA nucleotides. These chemical modifications are ubiquitous across all domains of life, with a vast range of functional and structural implications. To date, over 170 RNA modifications have been reported, many of which have unknown roles in cellular processes. While the study of RNA modifications has increased exponentially over the past 10 years, the authors have identified several challenges for the field:
- A lack of standardization in methodology, which hinders interdisciplinary communication of results and techniques.
- The absence of coordination between institutions that study RNA modifications.
___________________
4All authors contributed equally, and correspondence should be addressed to all of them.
- Insufficient and unaffordable chemical standards for validating experimental investigations.
- A lack of availability, sensitivity, multiplexing capability, quantitative accuracy, and/or sample throughput in current methods and technologies for mapping RNA modifications.
- No unified, comprehensive database for communicating, collating, and confirming the results of all epitranscriptomic studies.
- Limited resources for conveying the results and importance of RNA modifications to stakeholders outside of RNA biology.
In studying these problems, the authors have concluded that a dedicated body for studying epitranscriptomics is needed. We propose the creation of a Unified Epitranscriptomics Consortium (EpiC) to establish standardized practices in the field, from sample preparation to experimental design. EpiC would establish a unified reporting language to facilitate cross-field collaborations and interdisciplinary knowledge communication. Through these efforts, EpiC would curate and maintain a programmatically accessible database of RNA modification structure, function, and position data.
A successful EpiC would be an organization like the existing ENCODE and Ensembl projects, drawing funding and leadership from federal sources, while collecting, maintaining, and reporting comprehensive information on all known or newly discovered RNA modifications. To carry this out, EpiC would aim for the following goals (detailed in Figure F-2):
- Generate and make available an accessible and affordable library of chemical standards, ranging from individually modified nucleosides to modified oligos and even full-length transcripts.
- Support and organize the development and validation of new methods and technologies to identify and map RNA modifications.
- Standardize practices and protocols for detecting and mapping RNA modifications.
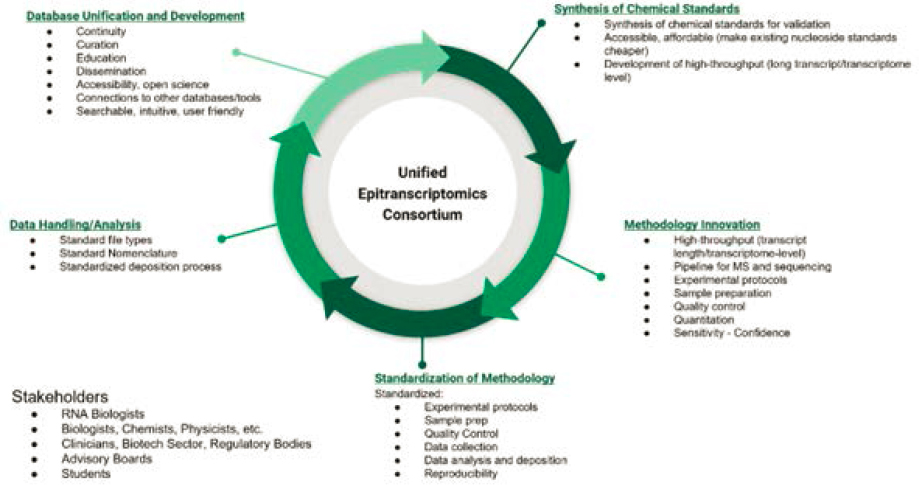
NOTES: The project is based on five continuous and interdependent components.
SOURCE: Originally created by the authors of this commissioned paper.
- Establish a syntax of RNA modifications, covering not only nomenclature but also data structures and file types.
- Maintain a centralized and highly curated database of RNA modifications and related information that is searchable, intuitive for nonexperts, highly extensible, and able to communicate with existing genomics databases such as the University of California, Santa Cruz (UCSC), Genome Browser, National Center for Biotechnology Information (NCBI) databases, and Ensembl, among others.
The creation of such a consortium would rapidly accelerate the field of epitranscriptomics, leading to significant improvements in our understanding of fundamental biological processes in addition to human diseases.
This paper discusses the challenges that epitranscriptomics researchers are currently faced with, both in wet-lab experiments and in computational approaches. We then describe our vision for the implementation and operation of EpiC and outline the main milestones expected within a 10-year horizon. We conclude by discussing the impact it will have to the scientific community and society at large.
Challenges to Mapping RNA Modifications
Experimental Challenges
Since the discovery of the first RNA modification, pseudouridine, more than 170 RNA modifications have been reported (Boccaletto et al., 2022), and the number is expected to rise. Following the discovery of RNA modifications, the field’s focus shifted to studying the effects of RNA modifications on RNA folding and stability. More recently, the roles of RNA modifications in cellular activities and their association with the development of diseases have drawn a lot of interest, especially after the interrelationships between writers, erasers, and readers of RNA modifications were demonstrated (Bokar et al., 1994; Jia et al., 2011). To further enhance understanding of the epitranscriptome, it is important to pinpoint the location of RNA modifications within each transcript. The ultimate goal is to create a map showing the distribution of all RNA modifications in any given transcriptome. The map of any epitranscriptome would be incomplete without information on how frequently a position is modified. Thus, from an analytical point of view, the basic requirements for mapping RNA modifications include the following:
- Identifying each RNA modification with high specificity and accuracy.
- Locating the position of RNA modification with single-nucleotide resolution.
- Determining how many copies (or what percentage) of the same transcript are modified at a given position.
Based on the analytical technique being used to generate the reporting signal of RNA modifications, the current methods for mapping RNA modifications can be divided into three groups, as shown in Figure F-3 (Yoluc et al., 2021).
Since in-depth reviews of the current methods are available (Felix et al., 2022; Moshitch-Moshkovitz, Dominissini, and Rechavi, 2022; Yoluc et al., 2021; Zhang, Lu, and Li, 2022), the remainder of this section discusses their applicability and technical challenges associated with using those methods to create a map of RNA modifications. Both mass spectrometry (MS) and nanopore-based methods can detect RNA modifications directly, and therefore are free from nonspecific antibody recognition, incompletion or errors from reverse transcriptase or ligase reactions, or cross-reactivity of some chemical reagents that are utilized in next-generation sequencing (NGS)
SOURCE: Originally created by the authors of this commissioned paper.
methods. This allows mass spectrometry– and nanopore-based methods to achieve a high level of accuracy. However, NGS methods can achieve significantly higher sensitivity and read longer transcripts. For all three groups of methods, chemical standards (e.g., pure oligos with known RNA sequences, modification(s), and/or concentrations) are required for proper calibration and data validation. However, chemical standards were largely missing in earlier reports using NGS, which could lead to unexpected errors and make it challenging to validate mapping information. For newer nanopore-based technology, the lack of chemical standards represents a challenge to evaluating the accuracy of quantitative measurements. While the chemical synthesis of RNA oligonucleotides with site-specific modifications has been established, the costs of customized RNA synthesis remain relatively high and therefore has not yet been utilized widely. Therefore, one of the key components of EpiC is supporting and coordinating the availability and accessibility of chemical standards. This will facilitate the development and validation of new or improved methods and technologies for mapping RNA modifications.
The mapping of only one type of RNA modification is far from sufficient since multiple different RNA modifications can be present in a given RNA transcript and each modification can alter RNA stability; folding; and/or interactions with other cellular components, including RNA-binding proteins (Chiu et al., 2021; Jones, Monroe, and Koutmou, 2020; Matsumura, Wei, and Sakai, 2023). In theory, mass spectrometry methods can be used to map most if not all the RNA modifications on a specific transcript (Wetzel and Limbach, 2016). Hence, the mass spectrometry methods are suitable for de novo or untargeted mapping of RNA modifications whose identity and position are unknown or unspecified. However, mass spectrometry methods require the purification of an individual transcript prior to top-down mass spectrometry measurements (Santos et al., 2022), or a specific group of transcripts has to be isolated before using the bottom-up MS approach (Lauman et al., 2023; Zhang et al., 2020). This means extra procedures and instrumentation are required. High-resolution mass spectrometry is required to pinpoint the exact location of RNA modifications within a given transcript. However, because of counterbalancing effects of mass resolution and measurement sensitivity, mass spectrometry methods end up being significantly less sensitive than NGS methods. The lower sensitivity in mass spectrometry measurements means fewer RNA ions are available, which in turn poses a challenge to attaining longer RNA sequence information from
the corresponding tandem mass spectrometry experiments. Further, in comparison with proteomic mass spectrometry, far fewer software programs are available to support the analysis of mass spectrometry data from RNA sequencing.
To the best of our knowledge, no current NGS method is designed to perform de novo or untargeted mapping. In other words, the identity of RNA modifications needs to be known and selected in each experiment prior to using current NGS methods to map RNA modifications (Cui et al., 2021; Finet, Yague-Sanz, and Hermand, 2022; Hu et al., 2022; Krogh and Nielsen, 2019; Lin et al., 2019; Thalalla Gamage et al., 2021). On the other hand, the higher sensitivity of NGS methods makes them more suitable for studies with limited amounts of RNA samples. Also, some of the NGS methods are available as commercial kits or fee-for-services. Because of the increasing availability of NGS instruments caused by the popularity of genome or transcriptome sequencing, setting up new mapping experiments has become less challenging. The main challenge for NGS methods is the limitation on multiplexing (e.g., mapping different RNA modifications in a single experiment). Although the mapping of different RNA modifications is feasible in a single sequencing experiment (Khoddami et al., 2019; Marchand et al., 2021), the total degree of multiplexing remains less competitive than the other methods. For instance, using nanopores to map RNA modifications at known positions, as many as 17 different RNA modifications were detected in the same experiment (Fleming et al., 2023). Besides higher multiplexing capability, the nanopore method has a unique benefit of being portable, thus allowing mapping experiments to be carried out anywhere at any time. This can be particularly useful for minimizing the errors from RNA degradation when storing biological samples is not an option.
Another consideration before selecting an appropriate method for mapping RNA modifications is the coexistence of many different transcripts in the same sample of interest. In specific cell types, thousands of genes were found to be expressed (Krjutskov et al., 2016), and the abundance of their corresponding transcripts differs vastly. The complexity of many RNA samples is further complicated by the presence of multiple RNA isoforms and/or partially degraded RNA molecules. For transcriptome-wide mapping, the challenge is the detection of modifications in low-abundance transcripts. To facilitate the detection of relatively low-abundance transcripts, one of the common practices in many sequencing protocols is removing ribosomal RNAs, which make up as much as 80 percent of total RNA in eukaryotes and even higher amounts in prokaryotic cells. If not removed, these abundant transcripts would either saturate the analytical platform or create high background noise. An additional approach to overcoming this challenge is targeted mapping, in which a specific transcript is selected and isolated from an RNA sample before mapping of RNA modifications. Although higher sensitivity can be achieved, the analytical challenges of targeted mapping include the inefficiency of isolating specific transcripts and the potential heterogeneity of the isolated transcripts that would require a separate mapping experiment for each transcript. Thus, targeted mapping would have a lower sample throughput than transcriptome-wide mapping. From the biological perspective, the challenge of targeted mapping is the selection and prioritization of transcripts that are related to the focus of a specific study.
Computational Challenges
With the rapid development of new instrumental techniques, including NGS and nanopore technologies to study these important biological changes to RNA, information is quickly building up. While several RNA modification databases have been created over the past decade, no truly comprehensive, centralized hub exists to upload, store, collate, and retrieve RNA modification datasets to advance the field of epitranscriptomics. Therefore, as a key component of the Unified Epitranscriptomics Consortium, we propose the development of an EpiC database. This database will act as a unified resource and data hub, enabling researchers across the globe to both contribute
and retrieve comprehensive information about all known or newly discovered RNA modifications—making it a primary resource for accessing epitranscriptome-wide RNA modification data.
To date, many RNA modification databases have been created to assist researchers in epitranscriptomics research. Modomics, RMBase (recently renamed ENCORE). and RNAMDB (Boccaletto et al., 2022; Cantara et al., 2011; Ma et al., 2022; Ramos, 2022; Xuan et al., 2018) are frequently referenced as the most comprehensive databases currently available in the field. Together, these repositories provide information about chemical structures and properties of many kinds of modified nucleosides, RNA modification sequence information, modification mappings from NGS, pathways of biosynthesis, phylogenetic sources, and links to human disease. Of these, Modomics seems to be the most inclusive, with information about more than 150 modifications available. In contrast, RMBase emphasizes more abundantly studied modifications, such as N6-methyladenosine (m6A), N1-methyladenosine (m1A), 5-methylcytosine (m5C), pseudouridine (Ψ), and ribose methylation (2′-O-Me), while still providing some information about other modified nucleotides (Boccaletto et al., 2018, 2022; Dunin-Horkawicz et al., 2006; Sun et al., 2016; Xuan et al., 2018). Still other repositories emphasize information about only one modification, such as MeT-DB (Liu et al., 2015, 2018), m6A-Atlas (Tang et al., 2021), m7GHub (Song et al., 2020), or REDIportal (Picardi et al., 2017), and newer repositories may focus on collecting data from only one methodology, such as the DirectRMDB database, which primarily incorporates nanopore data (Zhang et al., 2023). Many more databases exist that are not presented here but are discussed elsewhere (Ma et al., 2022; Ramos, 2022). Despite the long-standing operations of these databases, no single online utility exists that fulfills the vision of the proposed EpiC database to be the primary resource for storing RNA modification data on an epitranscriptome-wide scale.
This vision of the EpiC database is summarized in Figure F-4. The database should be a comprehensive data hub that is readily searchable to both advanced and nonexpert users. Information should encompass all forms of RNA modifications, across all types of RNA, and spanning all organisms regularly involved in epitranscriptomics research. The information should be presented in an approachable, easy-to-find format, with informative educational resources about RNA modifications and tutorials about how to effectively access the data within the EpiC database. It should also be interactive, and users should be able to visually adjust or manipulate both 2D and 3D mappings of genomic information or atomistic crystallographic structures. Data must be presented in a logical format, and users should be able to generate custom notebook pages that contain specific information they have selected from one or more EpiC webpages, therefore allowing them to organize and categorize information according to their research needs. Information kept within the database should be based on community-accepted standards, generated by standardized instrumental approaches, and carefully curated for accuracy by a consortium of experts. Finally, the database should not only act as a resource hub, but also contain powerful analytic tools built into the website to provide a digital workbench for scientific researchers. Thereby, investigators can directly evaluate specific information, perform new analyses, and accelerate epitranscriptomics discoveries within a common platform. For modifications that have known disease associations, these links should be established with clearly referenced publications. Together, these features will enable a more thorough mapping RNA modifications across the transcriptome within a common interface to connect researchers across the globe, helping to identify yet unknown links between epitranscriptomics, biology, and human health, while promoting cross-field collaborations and communications.
Many challenges exist in the creation of the EpiC database. To begin, there are many different types of RNA modifications, deriving from both reversible and unidirectional modification mechanisms, and information pertinent to each type of modification may differ by its underlying biochemistry. For example, a discussion of “erasers” would be irrelevant to the unidirectional A-to-I (Inosine) editing. Yet, both types of RNA modifications should be documented and deposited in the EpiC database. A current lack of standardization, data-file formats, and nomenclature

SOURCE: Originally created by the authors of this commissioned paper.
prevents consistency in data deposition, notation, and labeling, as well as the universal application of web-based analytic tools to all available or newly generated datasets. Vast amounts of information are already present in the literature, with more generated daily in research laboratories around the world. Retrieval and curation of this data requires a significant investment of personnel and resources to collect, sort, classify, and organize it into a centralized data hub. Finally, a dedicated team of software engineers and web developers, with skills that typically lie beyond the traditional capabilities of scientists in research labs, are needed to design the EpiC database platform and integrate both interactive visual tools and powerful analytic utilities within it. These capabilities will help researchers take full advantage of this centralized resource hub. Despite the tremendous obstacles present, successes in external repositories, such as the ENCODE project, provide confidence that a consortium of full-time staff and collaborative research lab members can successfully accomplish this task.
Management Plan
The EpiC project will be modeled after prior consortia of similar scope and size, such as the Human Genome Project, the ENCODE project, and the Extracellular RNA consortium. Given the need for materials and information sharing, at least two types of centralized facilities will be needed: one for the production and distribution of chemical standards, and one for the collection and dissemination of mapping information.
The facility that will synthesize and distribute chemical standards and other reagents should adopt production protocols that will be developed in the early phases of the project. Production will need to be scalable, and the facility should be organized to index and store synthetic RNA modification standards, as well as distribute them to production-phase laboratories that would request them. Indexing will be based on standardized nomenclature that will be agreed upon early in the progress of the project and should accommodate all possible RNA modifications, including ones available as modified RNA bases, nucleosides, nucleotides, and oligonucleotides. It should also provide the requesting laboratories with standardized detection protocols for each RNA modification.
The centralized information facility needs to have the capability to cope with an immense amount of incoming data. The data will need to be in a standardized electronic format or filetype. The data will need to undergo extensive quality control and quality assurance (QC/QA) procedures and become available to the scientific community and the public via dissemination to publicly available outlets, such as the UCSC Genome Browser, the NCBI databases, Ensembl, etc., as well as a separate website dedicated to the EpiC project. The information center will need to facilitate data exchange with other major databases so that it will act as the main resource hub for epitranscriptomics studies. Based on experience from the Human Genome Project, it will certainly be beneficial to many epitranscriptomic studies if regional core facilities for mapping RNA modifications are available, which can also be coordinated and managed by EpiC.
The EpiC-participating laboratories will benefit if housed within multidisciplinary research centers. Such centers will provide distinct capabilities, such as equipment and personnel sharing, as well as the integration of various disciplines and expertise needed to complete the task. Such disciplines include organic chemists, analytical chemists, sequencing experts, RNA biologists, bioinformaticians, software and database engineers, computer scientists, and server engineers. Thus, consortium partners should include laboratories that study RNA modifications in a variety of experimental samples, including model and non-model organisms, cell lines, and patients; laboratories that focus on the development of computational pipelines for data analysis; and industry partners, through the establishment of public–private partnerships. Given the complexity of the EpiC task, the laboratories participating in the production phase can also be distributed to include smaller research centers and individual laboratories, provided that they follow the same standardized data
generation procedures. This can be accomplished by training principal investigators and personnel from participating laboratories.
For the project to operate seamlessly, it should operate under the auspices of the National Institutes of Health, the National Science Foundation, the Department of Energy, and/or the Department of Defense. The lead agency (or agencies) will have the final word on funding and policy decisions and will be advised by the steering committee. The steering committee will be composed of scientists with expertise in epitranscriptomics that have a clear understanding of the project goals. Consistent with other consortia, a hierarchical structure supervised by a chair/co-chairs with a leadership team under them could provide a successful mechanism for efficient communication and direction. This committee’s responsibilities will include the following:
- Goal setting within the proposed timeline.
- Coordination and facilitation of the production-phase labs.
- Dissemination of data and ensuring accessibility of products.
- Quality control and assurance of experimental processes through the generation of a standardized peer review process for newly developed protocols.
- Reporting to funding agencies directly and to the scientific community through conference presentations and publications.
To ensure that EpiC goals are met, an External Advisory Board should be established. The board may include scientists that do not actively participate in the consortium but have expertise in the methodology and goals of EpiC. The main role of this board would be to provide feedback to the steering committee on EpiC’s progress and goals.
Additionally, to enhance productivity, synergy among labs, and the impact of the project, an Education Committee should be established. This committee will have dual objectives. It will be responsible for training prospective participating laboratories in standardized methods by organizing workshops and developing training materials. It will also be responsible for developing educational materials that will be adopted by undergraduate faculty to prepare students for the skills needed to join the workforce of the production phase labs. Other committees, such as ones that are focused on specific scientific tasks, equity and accessibility, etc. will be formed at the discretion of the steering committee.
Timeline
To adequately prepare for EpiC, it is imperative that clear timelines are established to ensure progress. These objectives are divided into near-, medium- and long-term goals, but it is important to note that many of these objectives will be overlapping and longitudinal. See Figure F-5.
Near-Term Goals (1–2 years)
- Establish a steering committee. The nomination and selection of the committee members with expertise in epitranscriptomes or related areas should be done as soon as possible, as this will allow for the other aspects of the project to be run in parallel. While rigorous vetting is necessary to ensure both competency and dedication, this process should be completed within the first few months of the inception of EpiC.
- Obtain feedback from existing/successful consortia. The success of other consortia, such as the Human Genome Project, ENCODE, and the Extracellular RNA Communication Consortium highlight both the feasibility and potential for success of the Unified Epitranscriptomics Consortium proposed here. Establishing communication with those and
- other consortia to identify successes and failures that they experienced throughout their processes would streamline EpiC for a more rapid return on investment. These lines of communication should be opened by the steering committee within the first few months of their leadership.
- Secure funding. Funding for EpiC will need to be secured from its onset. As mentioned above, several potential funding agencies have been identified. Irrespective of origin, the funding for EpiC needs to be secured quickly and will ideally be a renewable commitment to ensure long-term success. Years after EpiC’s establishment, discoveries, and successes, there will be continuous expenses in the form of administrative and scientific personnel, data upkeep and management, web server costs, and others. As such, securing long-term funding is critical.
- Hire personnel. EpiC will be a large undertaking and the hiring of personnel will reflect that potential. Hiring key personnel will occur following the establishment and guidance of the steering committee but will continue throughout the duration of EpiC. Among the first hires should be the administrative staff to maintain the consortium and the technological staff to begin development of the website and database. Computer scientists and database engineers should be considered a priority among personnel hires, as this will provide researchers with a resource to reference for their work and a location to deposit their findings.
Medium-Term Goals (2–5 years)
- Gather technical partners. Following establishment and initial internal hires, EpiC would need to look outwards and create a network of researchers who can address its goals. The generation of these partnerships could be aided by the creation of research grants, allocated through funding for EpiC, that would provide resources for collaborators to work towards projects that align with the defined objectives of the steering committee. This stage begins the true consortium aspect of EpiC.
- Standardize nomenclature and protocols. The lack of standardized nomenclature in epitranscriptomics can lead to difficult communication between researchers of different backgrounds. Therefore, agreeing upon unified terminology should be done early in the process, with the help of the steering committee and external scientists who are partnering with EpiC. At this stage, EpiC can begin to turn toward making technical advancements in the field. Similarly, experts should come to a consensus on optimized protocols that future researchers can use with confidence to ensure reproducibility among experimental results from mapping RNA modifications.
- Synthesis of chemical standards. In conjunction with standardized protocols, scientists need to obtain pure chemical standards for use in their experiments. By partnering with academic or industry laboratories that can produce chemical standards for RNA modifications, EpiC researchers can have confidence that their experiments utilize the proper controls and standards.
- Development of new methods and technologies. Innovative methods and technologies for mapping the complete set of RNA modifications with higher sensitivity, accuracy, and throughput are needed to advance the epitranscriptomic field. EpiC can play a role in coordinating academic and industrial efforts as well as organizing the testing of new methods or technologies.
- Build database and application programming interface (API). Having hired personnel early on to begin database creation, the database should be approaching operational levels at this stage. With that comes the necessity to create an interface with which all stakeholders
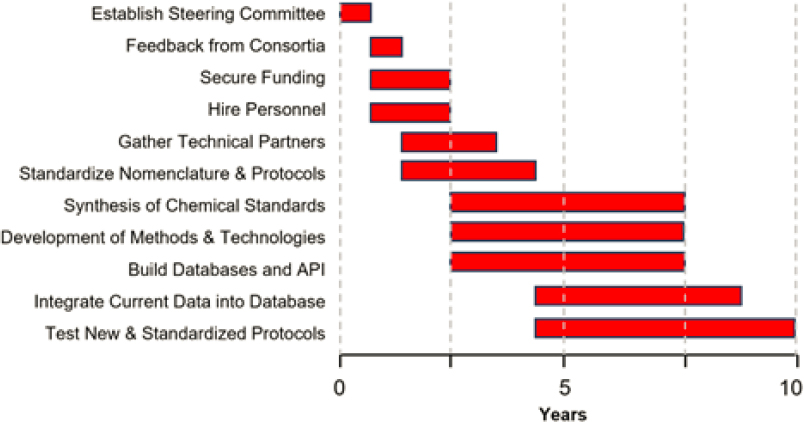
NOTES: The creation and maintenance of EpiC will be a major undertaking that will require large levels of support. Here we propose a realistic timeline for accomplishing milestones for EpiC over the next few years. API = application programming interface.
SOURCE: Originally created by the authors of this commissioned paper.
- can interact with the database to obtain their desired information. This will be facilitated through a Large Language Model that allows users to easily search for their information without needing to decipher technical jargon that could limit the accessibility of the database.
Long-Term Goals (5–10 years)
- Integrate current data into database. One challenge for current epitranscriptome researchers is that data and information are spread across numerous sites, many of which may not be curated or maintained. At the start of the long-term goals, EpiC will build upon the database established through the short- and medium-term goals, compiling all epitranscriptomic information spread across different sites into one central repository. This process will take a large effort, but it is essential that all relevant epitranscriptome information is housed in one location.
- Continue technological development. With the advancements of the medium-term goals of chemical standards and protocols, new discoveries will have been made in these areas which would require continued validation and refinement of approaches. This process will continue indefinitely as procedures are updated. This also provides opportunities for recruiting and training new scientists.
Impact of the EpiC Project
By establishing standards, mapping the epitranscriptome, exploring disease associations, and fostering collaboration, the EpiC project will significantly contribute to RNA biology and its translational applications. In particular, the creation of the EpiC database will impact the RNA community by providing an integrated resource of the different types of RNA modifications across all
species, tissues, transcripts, developmental stages, and disease states. Importantly, such a resource will facilitate the mapping of known and unknown RNA modifications across the transcriptome and the exploration of their functional relevance.
The datasets unified through the EpiC project could then be leveraged by the bioinformatics, data science, and artificial intelligence fields to develop novel algorithms for the detection of biological patterns and networks linked to RNA modification. Subsequently, this knowledge could be harnessed by the bioengineering and synthetic biology fields to develop novel biotechnology tools and processes. Moreover, the EpiC project will reveal biomarkers and pathways dependent upon RNA modification that could be targeted for diagnostic and therapeutic applications.
Notably, the EpiC project will impact the biomedical community by providing insight into undiscovered areas of human biology connected to epitranscriptomics. In turn, this would facilitate the identification of disease-causing genes and the molecular basis of complex pathologies such as multisystem disorders, neurodegeneration, and cancer. At the global level, the identification of functional epitranscriptomic patterns would open new areas of research for epidemiological studies. In summary, the achievements of the EpiC project are expected to impact multiple communities in the academic, engineering, and industry sectors that will benefit science and society.
REFERENCES
Boccaletto, P., M. A. Machnicka, E. Purta, P. Piatkowski, B. Baginski, T. K. Wirecki, V. de Crécy-Lagard, R. Ross, P. A. Limbach, A. Kotter, M. Helm, and J. M. Bujnicki. 2018. “MODOMICS: A database of RNA modification pathways. 2017 update.” Nucleic Acids Research 46(D1):D303–D7. https://doi.org/10.1093/nar/gkx1030.
Boccaletto, P., F. Stefaniak, A. Ray, A. Cappannini, S. Mukherjee, E. Purta, M. Kurkowska, N. Shirvanizadeh, E. Destefanis, P. Groza, G. Avşar, A. Romitelli, P. Pir, E. Dassi, S. G. Conticello, F. Aguilo, and J. M. Bujnicki. 2022. “MODOMICS: A database of RNA modification pathways. 2021 update.” Nucleic Acids Research 50 (D1): D231–D235. https://doi.org/10.1093/nar/gkab1083.
Bokar, J.A., M. E. Rath-Shambaugh, R. Ludwiczak, P. Narayan, and F. Rottman. 1994. “Characterization and partial purification of mRNA N6-adenosine methyltransferase from HeLa cell nuclei. Internal mRNA methylation requires a multisubunit complex.” Journal of Biological Chemistry 269(26):17697–17704.
Cantara, W. A., P. F. Crain, J. Rozenski, J. A. McCloskey, K. A. Harris, X. Zhang, F. A. Vendeix, D. Fabris, and P. F. Agris. 2011. “The RNA Modification Database, RNAMDB: 2011 update.” Nucleic Acids Research 39 (suppl_1): D195–201. https://doi.org/10.1093/nar/gkq1028.
Chiu, N. H. L., J. H. Simpson, H. Wang, and B. A. Tannous. 2021. “A theoretical perspective of the physical properties of different RNA modifications with respect to RNA duplexes.” BBA Advances 1:100025. https://doi.org/10.1016/j.bbadva.2021.100025.
Cui, J., Q. Liu, E. Sendinc, Y. Shi, and R. I. Gregory. 2021. “Nucleotide resolution profiling of m3C RNA modification by HAC-seq.” Nucleic Acids Research 49 (5): E27. https://doi.org/10.1093/nar/gkaa1186.
Dunin-Horkawicz, S., A. Czerwoniec, M. J. Gajda, M. Feder, H. Grosjean, and J. M. Bujnicki. 2006. “MODOMICS: A database of RNA modification pathways.” Nucleic Acids Research 34:D145–D149. https://doi.org/10.1093/nar/gkj084.
Felix, A. S., A. L. Quillin, S. Mousavi, and J. M. Heemstra. 2022. “Harnessing nature’s molecular recognition capabilities to map and study RNA modifications.” Accounts of Chemical Research 55(16):2271–2279. https://doi.org/10.1021/acs.accounts.2c00287.
Finet, O., C. Yague-Sanz, and D. Hermand. 2022. Epitranscriptomic mapping of RNA modifications at single-nucleotide resolution using rhodamine sequencing (Rho-seq). STAR Protocols 3(2):101369. https://doi.org/10.1016/j.xpro.2022.101369.
Fleming, A. M., P. Bommisetti, S. Xiao, V. Bandarian, and C. J. Burrows. 2023. “Direct nanopore sequencing for the 17 RNA modification types in 36 locations in the E. coli ribosome enables monitoring of stress-dependent changes.” ACS Chemical Biology 18(10):2211–2223. https://doi.org/10.1021/acschembio.3c00166.
Hu, L., S. Liu, Y. Peng, R. Ge, R. Su, C. Senevirathne, B. T. Harada, Q. Dai, J. Wei, L. Zhang, Z. Hao, L. Luo, H. Wang, Y. Wang, M. Luo, M. Chen, J. Chen, and C. He. 2022. “m6A RNA modifications are measured at single-base resolution across the mammalian transcriptome.” Nature Biotechnology 40 (8): 1210–1219. https://doi.org/10.1038/s41587-022-01243-z.
Jia, G., Y. Fu, X. Zhao, Q. Dai, G. Zheng, Y. Yang, C. Yi, T. Lindahl, T. Pan, Y. G. Yang, and C. He. 2011. “N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO.” Nature Chemical Biology 7 (12): 885–887. https://doi.org/10.1038/nchembio.687.
Jones, J. D., J. Monroe, and K. S. Koutmou. 2020. “A molecular-level perspective on the frequency, distribution, and consequences of messenger RNA modifications.” Wiley Interdisciplinary Reviews: RNA 11(4):E1586. https://doi.org/10.1002/wrna.1586.
Khoddami, V., A. Yerra, T. L. Mosbruger, A. M. Fleming, C. J. Burrows, and B. R. Cairns. 2019. “Transcriptome-wide profiling of multiple RNA modifications simultaneously at single-base resolution.” Proceedings of the National Academy of Sciences 116 (14): 6784–6789. https://doi.org/10.1073/pnas.1817334116.
Krjutskov, K., S. Katayama, M. Saare, M. Vera-Rodriguez, D. Lubenets, K. Samuel, T. Laisk-Podar, H. Teder, E. Einarsdottir, A. Sluments, and J. Kere. 2016. “Single-cell transcriptome analysis of endometrial tissue.” Human Reproduction 31(4):844–853. https://doi.org/10.1093/humrep/dew008.
Krogh, N., and H. Nielsen. 2019. “Sequencing-based methods for detection and quantitation of ribose methylations in RNA.” Methods 156:5–15. https://doi.org/10.1016/j.ymeth.2018.11.017.
Lauman, R., H. J. Kim, L. K. Pino, A. Scacchetti, Y. Xie, F. Robison, S. Sidoli, R. Bonasio, and B. A. Garcia. 2023. “Expanding the epitranscriptomic RNA sequencing and modification mapping mass spectrometry toolbox with field asymmetric waveform ion mobility and electrochemical elution liquid chromatography.” Analytical Chemistry 95(12):5187–5195. https://doi.org/10.1021/acs.analchem.2c04114.
Lin, S., Q. Liu, Y. Z. Jiang, and R. I. Gregory. 2019. “Nucleotide resolution profiling of m7G tRNA modification by TRACSeq.” Nature Protocols 14(11):3220–3242. https://doi.org/10.1038/s41596-019-0226-7.
Liu, H., M. A. Flores, J. Meng, L. Zhang, X. Zhao, M. K. Rao, Y. Chen, and Y. Huang. 2015. “MeT-DB: A database of transcriptome methylation in mammalian cells.” Nucleic Acids Research 43:D197–203. https://doi.org/10.1093/nar/gku1024.
Liu, H., H. Wang, Z. Wei, S. Zhang, G. Hua, S-W. Zhang, L. Zhang, S-J. Gao, J. Meng, X. Chen, and Y. Huang. 2018. “MeT-DB V2.0: Elucidating context-specific functions of N6-methyl-adenosine methyltranscriptome.” Nucleic Acids Research 46(D1):D281-D7. https://doi.org/10.1093/nar/gkx1080.
Ma, J., L. Zhang, S. Chen, and H. Liu. 2022. “A brief review of RNA modification related database resources.” Methods 203:342-353. https://doi.org/10.1016/j.ymeth.2021.03.003.
Marchand, V., V. Bourguignon-Igel, M. Helm, and Y. Motorin. 2021. “Mapping of 7-methylguanosine (m7G), 3-methyl-cytidine (m3C), dihydrouridine (D) and 5-hydroxycytidine (ho5C) RNA modifications by AlkAniline-Seq.” Methods in Enzymology 658:25–47. https://doi.org/10.1016/bs.mie.2021.06.001.
Matsumura, Y., F. Y. Wei, and J. Sakai. 2023. “Epitranscriptomics in metabolic disease.” Nature Metabolism 5(3):370–384. https://doi.org/10.1038/s42255-023-00764-4.
Moshitch-Moshkovitz, S., D. Dominissini, and G. Rechavi. 2022. “The epitranscriptome toolbox.” Cell 185(5):764–776. https://doi.org/10.1016/j.cell.2022.02.007.
Picardi, E., A. M. D’Erchia, C. L. Giudice, and G. Pesole. 2017. “REDIportal: A comprehensive database of A-to-I RNA editing events in humans.” Nucleic Acids Research 45(D1):D750-D7. https://doi.org/10.1093/nar/gkw767.
Ramos, J. 2022. “RNA modifications: An overview of select web-based tools.” RNA 28(11):1440–1445. https://doi.org/10.1261/rna.079443.122.
Santos, I. C., M. Lanzillotti, I. Shilov, M. Basanta-Sanchez, A. Roushan, R. Lawler, W. Tang, M. Bern, and J. S. Brodbelt. 2022. “Ultraviolet photodissociation and activated electron photodetachment mass spectrometry for top-down sequencing of modified oligoribonucleotides.” Journal of the American Society for Mass Spectrometry 33 (3): 510–520. https://doi.org/10.1021/jasms.1c00340.
Song, B., Y. Tang, K. Chen, Z. Wei, R. Rong, Z. Lu, J. Su, J. P. de Magalhães, D. J. Rigden, and J. Meng. 2020. “m7GHub: Deciphering the location, regulation and pathogenesis of internal mRNA N7-methylguanosine (m7G) sites in human.” Bioinformatics 36 (11): 3528–3536. https://doi.org/10.1093/bioinformatics/btaa178.
Sun, W. J., J-H. Li, S. Liu, J. Wu, H. Zhou, L-H Qu, and J-H. Yang. 2016. “RMBase: A resource for decoding the landscape of RNA modifications from high-throughput sequencing data.” Nucleic Acids Research 44(D1):D259-65. https://doi.org/10.1093/nar/gkv1036.
Tang, Y., K. Chen, B. Song, J. Ma, X. Wu, Q. Xu, Z. Wei, J. Su, G. Liu, R. Rong, Z. Lu, João P. de Magalhães, D. J. Rigden, and J. Meng. 2021. “m6A-Atlas: A comprehensive knowledgebase for unraveling the N6-methyladenosine (m6A) epitranscriptome.” Nucleic Acids Research 49 (D1): D134–D143. https://doi.org/10.1093/nar/gkaa692.
Thalalla Gamage, S., A. Sas-Chen, S. Schwartz, and J. L. Meier. 2021. “Quantitative nucleotide resolution profiling of RNA cytidine acetylation by ac4C-seq.” Nature Protocols 16(4):2286–2307. https://doi.org/10.1038/s41596-021-00501-9.
Wetzel, C., and P. A. Limbach. 2016. “Mass spectrometry of modified RNAs: Recent developments.” The Analyst 141(1):16–23. https://doi.org/10.1039/c5an01797a.
Xuan, J-J., W-J. Sun, P-H. Lin, K-R. Zhou, S. Liu, L-L. Zheng, L-H. Qu, and J-H. Yang. 2018. “RMBase v2.0: Deciphering the map of RNA modifications from epitranscriptome sequencing data.” Nucleic Acids Research 46(D1):D327–D34. https://doi.org/10.1093/nar/gkx934.
Yoluc, Y., G. Ammann, P. Barraud, M. Jora, P. A. Limbach, Y. Motorin, V. Marchand, C. Tisné, K. Borland, and S. Keller. 2021. “Instrumental analysis of RNA modifications.” Critical Reviews in Biochemistry and Molecular Biology 56(2):178–204. https://doi.org/10.1080/10409238.2021.1887807.
Zhang, N., S. Shi, X. Wang, W. Ni, X. Yuan, J. Duan, T. Z. Jia, B. Yoo, A. Ziegler, J. J. Russo, W. Li, and S. Zhang. 2020. “Direct sequencing of tRNA by 2D-HELS-AA MS Seq reveals its different isoforms and dynamic base modifications.” ACS Chemical Biology 15 (6): 1464–1472. https://doi.org/10.1021/acschembio.0c00119.
Zhang, Y., J. Jiang, J. Ma, Z. Wei, Y. Wang, B. Song, J. Meng, G. Jia, J. P. de Magalhães, D. J Rigden, D. Hang, and K. Chen. 2023. DirectRMDB: A database of post-transcriptional RNA modifications unveiled from direct RNA sequencing technology. Nucleic Acids Research 51(D1):D106–D116. https://doi.org/10.1093/nar/gkac1061.
Zhang, Y., L. Lu, and X. Li. 2022. “Detection technologies for RNA modifications.” Experimental & Molecular Medicine 54(10):1601–1616. https://doi.org/10.1038/s12276-022-00821-0.
TOWARDS THE COMPREHENSIVE MAPPING OF THE HUMAN EPITRANSCRIPTOME
Author List
David G. Courtney, Queen’s University Belfast
Dimitri Pestov, Rowan University
Margot Martinez-Moreno, Brown University
Context
Advancements in technology have significantly expanded knowledge of RNA modifications, providing new insights into their potential functions and relevance to human diseases. There has also been substantial growth in the awareness of the multifaceted control these modifications exert over various aspects of RNA biology, including processing, stability, localization, and translation of mRNA into functional proteins. Nevertheless, it is important to recognize that the study of RNA modifications is still in its early stages because of their formidable complexity and dynamic nature. More than 150 chemical alterations of nucleobases have been identified in both protein-coding and noncoding RNA across different organisms. Deciphering the regulatory roles of these modifications is particularly challenging given their intricate cell type– and context-dependent patterns.
The Problem
Despite notable strides made by research groups worldwide in developing new methods and analyzing the biological functions of RNA modifications, the overall research effort in this field remains fragmented. Additionally, the lack of effective communication and data integration among research groups continues to hinder progress. Therefore, we believe there is a pressing need for the development of a comprehensive and collaborative platform to advance our understanding of the epitranscriptome, while facilitating data exchange and promoting standardization practices across the scientific community.
The Solution
We propose the formation of a global consortium consisting of public research institutions and industry groups. This consortium would serve as a centralised hub for facilitating the development of RNA modification mapping technologies and depositing and sharing information, while promoting consistent standards and data formats that can be used interchangeably by researchers interested in various organisms or tissues. The proposed consortium will encourage researchers employing various technologies to work together effectively. In addition, it will provide resources and infrastructure and facilitate reliable and consistent data analysis, thereby enabling comprehensive and rigorous research in the epitranscriptomics field.
Why Is This Important?
At this point in time the unknowns in the field of epitranscriptomics are vast. Scientists lack a comprehensive and robust reference epitranscriptome, similar in a way to what scientists faced in 1990 with the absence of a reference human genome. The ultimate aim of the proposed consortium will be the establishment of a reference human epitranscriptome that would provide researchers worldwide with important information regarding each nucleoside of every known RNA, including mRNA and all noncoding RNAs. We envisage that this dataset would have wide-ranging implications in the fields of cancer biology, microbiology, diagnostics, and RNA biology. For instance, it is well known that cancerous cells have a distinctly altered epitranscriptome when compared with wild-type cells of the same tissue (Baylin and Jones, 2016; Janin, Davalos, and Esteller, 2023; Miano et al., 2021), while almost every virus that has been investigated has been shown to acquire epitranscriptomic marks during replication (Netzband and Pager, 2020; Tsai and Cullen, 2020; Yang, Zhao, and Zhang, 2023). RNA modifications have also been implicated in “disguising” foreign RNA from innate immune sensors, a property most notably utilized in the recent SARS-CoV-2 mRNA vaccines (Morais, Adachi, and Yu, 2021). A comprehensive map of human RNA modifications could potentially lay the groundwork for a wide range of translational research outputs. As one example, these could include the development of modification inhibitors that could either become a part of new chemotherapeutic regimes for cancers or act as a new generation of broad-spectrum antivirals (Liu et al., 2022, 2023; Sun et al., 2023; Zhang et al., 2022).
Lessons from the Past: The Human Genome Project and Its Impact on Science and Medicine
In the mid-80s, because of the emerging roles and technologies related to DNA sequencing, recombinant DNA technologies, etc., a meeting was held in Alta, Utah, with one goal in mind: uncover methods on detection of DNA variants in the context of the Hiroshima-Nagasaki survivors (Cook-Deegan, 1989; Delehanty, White, and Mendelsohn, 1989). The principal conclusion of that meeting was that, without a reference database, the existing methods were just insufficient for measuring variants with enough sensitivity. This led to the formation of a human genome workshop that, in 1986, started setting up the outline of what such a project would entail and to assess its feasibility (CSHL, 1989). The final council was formed in 1988 with the formation of HUGO, the Human Genome Organization (Liu, 2009) the International Human Genome Sequencing Consortium was established. The Human Genome Project (HGP) started in 1990 and stands as one of the most remarkable scientific enterprises around the turn of the century. The objective was to sequence and ensemble the human genomic DNA sequence, or roughly 3 billion DNA base pairs. This ambitious project involved the coordination of scientists, researchers, institutions, and companies from all over the world that not only sequenced and assembled the DNA but also developed innovative technologies and computational methods that allowed for analysis and interpretation of the vast amount of generated genetic data. In 1998, Celera Genomics started competing with the HGP to win the race on who would finish the mapping of the human genomic DNA first, although they ended up joining forces and finished the first draft in 2000. A year later, the draft was published in Nature (Lander et al., 2001) and Science (Venter et al., 2001), finishing the complete sequence in 2003, just in time for the 50th celebration of the discovery of the double helix of DNA by Watson and Crick.
The HGP had far-reaching implications for society in general and medicine in particular. The most immediate contribution was to medical research, not only serving its original purpose as a “template” of all the human genes, but to the development of personalized medicine (Gibbs, 2020). It was also a boon to subsequent projects that involved data sharing, computerized advances and the need of database creation: HapMap Project, the 1000 Genomes Project, The Cancer Genome
Atlas Program (TCGA), National Center for Biotechnology Information (NCBI), etc. It also pushed the boundaries of DNA sequencing technology: the development of new methods led to relatively cheap, universal, and accessible sequencing technologies; today, human genome sequences cost less than $1,000 and the waiting time for results can be a few weeks or even days. This has subsequently accelerated progress in various fields of biological and biomedical research. Also, the unprecedented amount of available genetic information paved the way for computational biologists to thrive. Moreover, DNA profiling and forensic applications that have emerged after the HGP completion have revolutionized forensic sciences. One of the most remarkable aspects of the HGP is that it demonstrated the power of international collaboration and open science.
By looking back at the context of that summit in 1984, we can draw parallels with the needs in the epitranscriptomic field today: in order to facilitate a deeper understanding of the epitranscriptome, we need to have a reference human epitranscriptome comparable to the reference human genome.
Near-Term Goals (1–3 Years)
- The first phase of establishing the consortium should begin with a founding meeting of a steering committee composed of academic researchers and, potentially, industry representatives. The purpose of this meeting is to define the initial scope of the consortium’s activities and identify stakeholders.
- Considering the vast amount of data present at the epitranscriptomic level, the best path forward may be to begin by examining one or a few well-defined human experimental cell systems, with a potential supplemental use of a simpler model system (such as yeast) for methodology development.
- To develop the workflow for comprehensive human epitranscriptome analysis, several key steps must be taken. First, standards must be established for sample collection and processing to ensure uniformity across different research groups. Second, data standards have to be devised to facilitate effective communication and data integration among researchers utilizing different methodologies. Third, rigorous analysis pipelines will need to be defined to ensure reliable and consistent data analysis. Fourth, development and establishment of a reference biochemical approach to map each type of modification will need to be done with sufficient sensitivity and reproducibility to identify the specific modifications present in the samples. Fifth, in order to allow for a more comprehensive understanding of the role of different modifications, functional assays have to be developed to quantify the biological impact of the modifications.
- A comprehensive database has to be established for the storage and sharing of the data obtained from different research groups. The database’s core structure and features need to accommodate the highly dynamic nature of the epitranscriptome. By their nature, RNA modifications are capable of rapidly adjusting how individual RNA molecules are utilized within cells, operating on much shorter timescales than conventional genetic or epigenetic regulatory mechanisms. For instance, RNA modifications could fine-tune gene expression programs that are only activated during specific stimuli, such as stress responses. In addition, RNA modifications may govern the differential utilization and fate of RNA molecules based on the cell’s microenvironment or even within subcellular regions, like translation and targeting to specific locations. To comprehensively understand RNA modifications, it is therefore essential to establish consistent standards regarding the biological samples that serve as the data source. Ideally, the metadata associated with a biological source being analysed should include a “4D” framework, encompassing detailed information about both the spatial and temporal context.
- Bioinformatic tools must be developed to integrate data from the different sources and to allow for the comparative analysis of different experiments. A scalable computational infrastructure should be established from the outset, involving core computational groups with expertise in emerging machine learning and artificial intelligence (AI) technologies to support wet labs participating in the consortium.
- The consortium must create chemical and RNA standards, as well as reference spike-ins, for developing new methods and validating pipelines that detect modified nucleotides. These resources should be made available to participating laboratories upon request, including those that join the project at a later stage.
Long-Term Goals
Once the analytical pipeline is optimised and considered robust and reliable, the consortium will identify the next steps to achieve the following long-term goals (see Figure F-6):
- Present a comprehensive epitranscriptome for major modifications on yeast (or another model organism) RNA in a publicly accessible database. This will provide proof-of-principle (in a similar vein to the Human Genome Project) that the methodologies are truly robust and reproducible across various labs.
- Follow this up with the publication of a comprehensive epitranscriptome of human RNA for some well-defined cell systems that were discussed and specified through completion of the near-term goals. This will initially take the form of, likely m6A, m5C and pseudouridine maps only for the defined cell system.
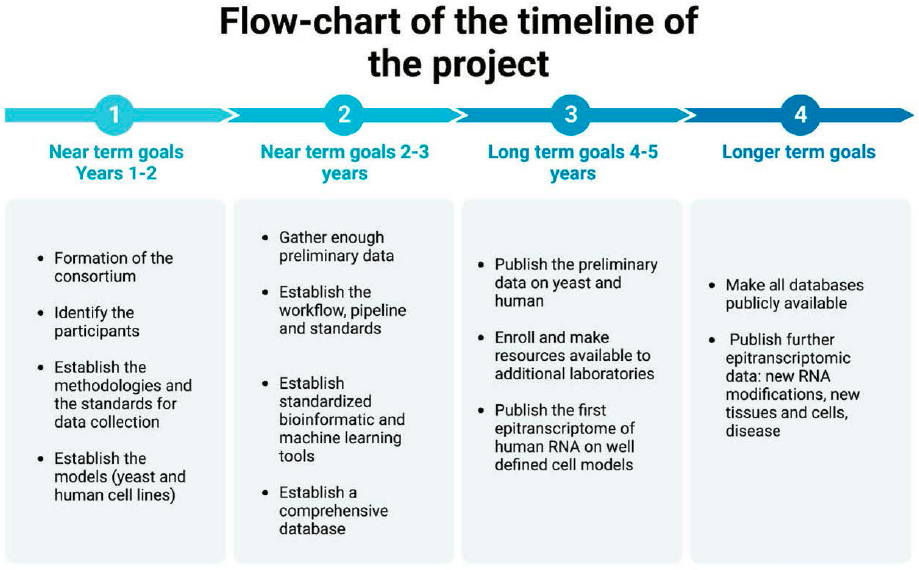
SOURCE: Originally created by the authors of this commissioned paper.
- Further epitranscriptomic mapping data will be published for additional modifications, including 2’Ome, ac4C, m1A and m7G, etc., will be published for human RNA from the same defined cell systems.
- With the establishment of a reproducible, robust, and sensitive mapping pipeline for a range of modifications, and the first publication of a comprehensive human epitranscriptome, the steering committee should meet again to discuss branching out to more biologically relevant cell systems such as primary cell cultures, organoids, or human tissues, and to explore different disease models such as cancer, immunological, or neurodegenerative conditions. At the same time, they may wish to explore how external stimuli alter the human epitranscriptome, such as drug treatments, cellular stress, infection, etc. We would hope that as these pipelines are more readily employed, answering these kinds of questions should become relatively straightforward and commonplace for any labs around the world that wish to become involved.
End Goal and Metrics
Overall, the best goal for the consortium at the initial stages of the project may be to establish a database of the human epitranscriptome for a single-cell system for as many modifications as possible. As a result, we envisage that this foundational effort would be scalable and expandable in all following phases of the project to cover any organisms/tissues beyond simply focusing on model human RNA. By collaborating and sharing standardized protocols, pipelines, and resources, this can help cut costs to individual laboratories while maintaining the quality and integrity of the research. Therefore, the objectives are twofold: an initial human epitranscriptome will be generated and published as a publicly available resource; and standardized protocols will be established and distributed for any researchers wishing to map RNA modifications in their cell system of choice.
The success of this project will be determined by the number of RNA modifications that standardized protocols can be established for, and the number of human cell systems that can have their epitranscriptomes mapped by the team within the lifetime of the project. It should not be overlooked that simply the establishment and publication of standardized and robust methods for the mapping of individual RNA modifications, which can be taken up by any researchers interested in epitranscriptomics, would be a worthy goal for the consortium and would have far reaching implications within the field.
Current Technologies and Methodologies
We are proposing using a combination of validated biochemical methodologies to map individual RNA modifications at single nucleotide resolution. The two predominant methods for single nucleotide resolution mapping are shown in Figure F-7 (Courtney, 2021). At present, numerous methods have been published for the mapping of modifications on mRNA including MAZTER-seq, bisulfite-sequencing, CMC-seq, Pseudo-seq, BID-seq, Nm-seq, RiboMethSeq, RedaC:T-seq, and AlkAniline-seq.
A novel method for mapping m6A modifications, known as MAZTER-seq, is a potential option for single nucleotide resolution of m6A across the transcriptome (Garcia-Campos et al., 2019). This method employs a bacterial endonuclease that will cleave 5’ of an ACA motif but is unable to cleave this motif when the first residue is an m6A. This enzyme has been coupled with a next-generation sequencing (NGS) library preparation protocol to allow for the de novo detection of m6A sites across eukaryotic RNA. What’s more, with adequate standards this method can also reveal the modification occupancy rate at a given site. Unfortunately, one caveat to this method is that it
SOURCE: Modified from Courtney, 2021.
will only identify roughly one quarter of m6A sites as it is only functional at identifying modified residues within an m6ACA motif.
An additional biochemical method that has been reported for modification mapping is RNA bisulfite sequencing. This method can specifically be used to uncover individual m5C modifications on cellular RNAs, and to quantify the occupancy rate. In the presence of sodium bisulfite, RNA is denatured at high temperatures to chemically deaminate unmethylated cytosine residues to uracil (Schaefer, 2009). m5C modified residues remain as unconverted cytosines, which is due to the low reactivity of m5C with hydrogen sulfite. A cytosine residue that remains, and thus was ‘protected’ from deamination, can then be detected by NGS and would be called as an m5C. One shortcoming of this method is the false reporting of additional modifications, namely 5-hydroxymethylcytidine and 5-formycytidine (f5C), which can also lead to protection from deamination. In addition, bisulfite conversion is affected by complex RNA secondary structure, with more complex structures drastically reducing efficiency. Therefore, adequate controls will have to be developed to be certain of calling a true m5C site.
The CMC-seq and pseudo-seq methods have been well-utilized at this stage to map ψ residues on eukaryotic RNA (Carlile et al., 2014; Schwartz et al., 2014). This protocol uses N-cyclohexyl-N′(2-morpholinoethyl) carbodiimide metho-p-toluenesulfonate (CMC) to modify only pseudouridinylated residues. The oversized CMC adduct on the ψ results in a complete block to reverse transcription and these prematurely stopped cDNA fragments can then be identified through NGS and
bioinformatic analysis (see Figure F-7). A control sample, consisting of the same RNA but in the absence of CMC is processed in parallel to set background levels of premature reverse transcriptase (RT) stopping.
Recently, two groups independently published very similar methods adapting conventional RNA bisulfite sequencing to a neutral pH to specifically map Ψ through inducing an RT deletion signature when a Ψ modification is present (Zhang, Dai, and He, 2023; Zhang et al., 2023). Again, through use of NGS, this deletion signature at a single uridine site can easily be identified when coupled with bioinformatic analyses. These methods allow for the determination and quantification of individual Ψ sites on cellular RNA with a relatively low read depth. But, as these methods are only recently published, adequate testing and optimization must be performed to determine the reliability of these methods before being employed by the consortium.
Two methods to accurately map the location of 2'Ome base modifications are RiboMethSeq (Marchand et al., 2016) and Nm-seq (Hsu et al., 2018). RiboMethSeq employs alkaline fragmentation on an RNA pool, where 2'Ome residues are generally resistant to fragmentation. These fragmented RNA are then ligated to adapters and processed for NGS. If sufficient sequencing depth is achieved, sites of fragmentation that are underrepresented in respect to the surrounding residues can be uncovered bioinformatically. It can then be assumed that the underrepresentation of these sites is caused by the presence of 2'Ome residues (Marchand et al., 2016). Of course, this results in a single-nucleotide resolution map of all 2'Ome sites, as well as the occupancy rate at individual sites. An alternative method to map 2'Ome base modifications is Nm-seq. This technique is more time consuming but has the benefit of requiring lower read depth. In Nm-seq, a series of oxidation–elimination–dephosphorylation reactions are performed on fragmented RNA from a given host. Through these cycles unmodified nucleotides are eliminated from the 3'UTR of the fragmented RNA until, eventually, a 2'Ome modified nucleotide is the 3' most residue. This modified residue is protected and thus remains in place through further cycles. This technique is then combined with NGS (Hsu et al., 2018). With this method, 2'Ome residues can be identified in a similar way to RiboMethSeq, with deep sequencing followed by bioinformatic analysis. While in RiboMethSeq, the researcher uncovers underrepresented sites at the periphery of the read indicating the presence of 2'Ome. Nm-seq relies on the identification of overrepresented sites at the 3'UTR to call a 2'Ome modification.
A biochemical method, termed RedaC:T-seq, has been reported for the single nucleotide mapping of ac4C modified residues on cellular RNA (Sturgill, Arango, and Oberdoerffer, 2022). This method makes use of the ability of sodium borohydride to specifically reduce ac4C residues, and through RT, this site is converted to a thymidine. This induced polymorphism at the site of ac4C modification can then be readily identified by NGS and straightforward bioinformatic analysis.
Finally, there are a few mapping methods that allow for the identification of multiple modifications at once. One of these is termed AlkAniline-seq (Marchand et al., 2018). This method is primarily for the biochemical identification of internal m7G modifications, but it can also be used to reveal the much rarer 3-methylcytidine and D modifications on cellular RNA. In short, this method involves the alkaline hydrolysis of purified RNA, followed by dephosphorylation and then aniline cleavage of abasic residues. NGS library preparation can then be performed, as the resulting 5'phosphates are ligatable with standard library adapters. The resulting libraries will be biased at the 5’ end towards the modification +1 position and can easily be determined bioinformatically.
In addition to those described above, additional methods for the mapping of ac4C (Arango et al., 2018; Sas-Chen et al., 2020; Thalalla Gamage et al., 2021), m7G (Zhang et al., 2019) and m1A (Zhou et al., 2019) have been published and could be explored further by this consortium.
However, the vast majority of these methods are relatively new and their robustness and reproducibility from lab to lab is yet to be tested experimentally. We believe that optimal biochemical methods for mapping common RNA modifications such as m6A, m5C pseudouridine and 2'Ome likely already exist, but that it will be for the consortium to test, validate, and demonstrate
experimentally which single method for each modification is reproducible worldwide. In addition to these approaches, we propose a secondary goal of establishing nanopore sequencing for modification calling, discussed further below.
Emerging Technologies and Methodologies
Though direct RNA sequencing through nanopore-based technology has been around for a number of years, Oxford Nanopore’s exciting new development of an RNA-specific pore is likely to revolutionize the sequencing of the epitranscriptome in the coming years. Previously, all direct RNA sequencing had been performed on DNA sequencing pores that had performed adequately but were not wholly fit-for-purpose. Through advances in the chemistry surrounding nanopore sequencing, an RNA-specific sequencing platform will become available in late 2023. This technology will greatly increase the sensitivity of detection for individual nucleotides. When this is coupled with a modification-aware basecaller, our ability to quickly and robustly “call” and quantify the modification rate of individual sites within cellular RNA could become relatively straightforward (Lucas and Novoa, 2023). Therefore, further research developing modification-aware basecallers trained on reads of transcripts containing a variety of RNA modifications at various stoichiometries and sequenced on the new RNA-specific nanopores is vitally important. Nanopore-based sequencing is already a powerful tool for mapping RNA modifications on native RNA (Fleming et al., 2023), and with the consortium leading further advances, as discussed, this could quickly become the all-in-one epitranscriptomic mapping tool the field needs. In the long-term, these emerging technologies will bring down the cost and technical ability required to map the cellular epitranscriptome while also making it a tool available to most labs across the world.
Single-cell RNA-seq is an ever-growing area of interest to RNA biologists, with the power it provides to identify cell-to-cell variations in transcript levels, splicing, and poly(A) tail length, where previously we could only measure these at a population-wide level (Arzallus-Luque and Conesa, 2018). With the establishment of robust biochemical mapping techniques for individual RNA modifications through this consortium, it is entirely realistic that some of these could be combined with current single-cell RNA sequencing methodologies to allow researchers to quantify nuanced differences in the epitranscriptomes of individual cells. In addition, single-cell RNA sequencing is already possible with nanopore sequencing (Lebrigand et al., 2020). However, this technique currently utilizes cDNA libraries that would thus remove any information regarding the modification status of individual nucleotides. It is not unreasonable to suggest that a single-cell direct RNA sequencing approach could also be on the horizon, using individual cell-specific bar codes ligated onto all transcripts from a given cell before sample preparation and nanopore sequencing. Therefore, if a demonstrably accurate and highly sensitive modification-aware basecaller, as described above, can be developed for direct RNA nanopore sequencing, then the mapping of single-cell epitranscriptomes could very easily be the next domino to fall within the fast-paced field of epitranscriptomic research.
Spatial transcriptomics, with other spatial technologies, has enabled scientists to dissect the organization and interaction of different cell types within tissue and its relationship with the microenvironment (Anderson et al., 2022; Hunter et al., 2021). It has also been argued that the main RNA modifications (m6A, m1A, etc.) constitute critical mechanisms of epigenetic regulation in immune response and tumorigenesis, also known as tumour microenvironment (Chen et al., 2021). Current techniques use spatially barcoded probes to detect and measure mRNA from tissue, while counterstaining with histological markers and antibodies. However, to the best of our knowledge, no epigenetics or RNA modifications can be detected. Spatial transcriptomics for RNA modifications may be, then, a useful tool to uncover the differential epigenetic modifications and to link them to the tumour microenvironment. While direct RNA mapping (as described above) could easily
be utilized to sequence spatially barcoded RNA transcripts, other modifications of the technique for mapping modifications may include the use of m6A antibodies, m6A PLA, and m6A seqFISH coupled to spatial transcriptomics.
Disciplines and Expertise Areas to be Engaged
This project should involve the collaboration of experts from diverse fields. International collaboration will be crucial. The research community will be any researchers worldwide who are interested in exploring modifications located on any transcript of interest. We envision the mapping of the RNA human transcriptome as an international scientific research effort that involves a wide range of key disciplines and expertise areas, which may include, but will not to be limited to the following:
- Biochemistry and molecular biology
- Genetics and epigenetics
- Synthetic chemistry
- Genomics
- Sequencing technology
- Computational biology and bioinformatics
- Structural biology
- Cell biology
- Biotechnology
- Robotics and automation
- Machine learning (ML) and artificial intelligence (AI)
Stakeholders
Since this is a large-scale international endeavour, a wide range of stakeholders from different sectors will need to be involved. These stakeholders will play an important role in various aspects of the project, as described further below:
- Government agencies. Typically, government agencies provide significant funding for this type of large-scale project. We envisage that the principal funding agencies should be international public funders, such as the National Institutes of Health or Department of Energy for the United States, and the United Kingdom Research and Innovation (UKRI) or European Research Council for Europe, at least for the first stages of the project.
- Research institutions and universities. Many research institutions and universities around the world may participate in scientific research, data generation, and analysis.
- Scientific community. As explained in the above section, scientists from various fields may actively contribute their expertise to the project.
- Private companies. Biotechnology, AI, pharmaceutical, and other types of companies may play a role in the project at later stages, as long as we ensure the freedom of the data (considerations explained below). These companies may be involved in the generation of new technologies as well as data acquisition and prediction systems.
- Non-profit organizations. Funding may come from private funds and trusts.
- Ethical, legal, and social implications (ELSI) committees. ELSI committees will be established by the consortium to identify and address the ethical, legal, and social implications of the project and its impact and will create and enforce the application of guidelines and regulations.
- General public. Over the years, the research and data generated by the project will have a tremendous impact for society, medicine, and ethics. Public engagement and education will be important aspects of the project.
- Media. A communications agency may be created to ensure the successful dissemination of the data and information on the project to the different stakeholders, agencies, and the general public.
- Policy makers. Toward the ending stages, policy makers and governmental agencies will help the ELSI committee shape the regulatory environment and policies related to the research and data sharing.
- Patients and patient advocacy groups. These groups may be interested in the potential medical applications of the epigenetic landscape discoveries and may discuss how to ethically use the data related to patients.
Workforce, Educational, or Societal Considerations
The consortium should be open to academic–private relationships. However, it is crucial that the resulting data remain a freely available public resource to which all researchers will have access. Ultimately, the overarching aim of this project would be to establish a publicly accessible database that can be accessed by anyone in the world at no cost. Free data sharing between scientists will help disseminate knowledge and drive down expenses by avoiding the generation of redundant data and its analysis. In addition, by promoting method development, providing a shared repository of referenced samples and standards, and creating database infrastructure, the consortium is anticipated to reduce research costs in a similar way as witnessed with DNA sequencing costs after the HGP (Hood and Rowan, 2013).
As the project builds knowledge on how the epitranscriptome shapes the susceptibility to disease, ethical questions may arise surrounding genetic and epigenetic testing, data privacy, and the potential misuse of data by health insurance companies and employers. These advances must be used only to explore the intricate mechanisms underlying health and disease in order to find cures, not to allow discrimination and promote social exclusion. The repercussions of this project may also impact and shape the access to medicine. As we discussed above, since the most significant impact of the mapping of the epitranscriptome would be its contribution to medical research and personalized medicine, some complex ethical questions surrounding epigenetic testing may arise. First, even with the mapping of the RNA modifications, every patient has a unique epigenetic landscape that may affect epitranscriptomics (Rasool et al., 2015). Even though the technologies will become cheaper and more accessible, a complete epigenetic panel may still be expensive for certain populations and for certain countries, which may lead to social exclusion and lack of accessibility in developed countries. In countries under development, patients will not be able to access these new resources.
Impact
We expect that this project would have an impact on multiple fields:
- Medicine. The comprehensive mapping of the human epitranscriptome, including the full diversity of RNA modifications, will have a tremendous impact on the medical and biomedical fields. RNA modifications have emerged as critical regulators of diverse cellular and biological processes, including development, differentiation, and disease origin and progression (Chen et al., 2021). By mapping all RNA modifications, the consortium will lay the foundation for researchers to understand the role of specific modifications
- (by organism, by tissue, by cell, and by cellular state) and their functional implications in disease. By understanding individual RNA modifications and their profiles in disease, researchers may be able to better predict their response to specific treatments or even tailor therapies based on their epigenetic signatures. This has the potential to profoundly impact medicine by enabling the development of new diagnostics and therapeutics, facilitating drug discovery, and contributing to personalised medicine approaches.
- Therapies/vaccines. With the ever-increasing importance of mRNA-based therapeutics, particularly insofar as the development of the next generation of vaccines, research into RNA modifications will be critical. The COVID-19 mRNA vaccines contain a large number of N1-methylpseudouridine (m1Ψ) modifications, selected specifically to reduce the immunogenicity of the foreign mRNA being delivered (Nance and Meyer, 2021). This builds off work originally pioneered by Dr. Karikó and colleagues, who demonstrated that particular RNA modifications, already found on cellular RNAs, can reduce the antagonism of the innate immune response when incorporated into in vitro transcribed RNA prior to transfection (Karikó et al., 2005). Through our evolving understanding of the biology surrounding RNA modifications, as laid out herein, we increase the likelihood of identifying other novel traits for RNA modifications that could be incorporated into the next round of mRNA-based therapeutics. For example, if a specific RNA modification within the 5’ UTR of an mRNA could increase translation efficiency, or a different RNA modification in the 3’ UTR slowed RNA degradation, then by incorporation of these modifications, a lower dose of mRNA could be utilized to yield the same expression. Thus, mRNA vaccines and therapeutics could be produced at a relatively faster rate. Any options to reduce the dose and increase efficacy of mRNA therapeutics will have a serious impact on humanity moving forward as our therapeutic regimes become more inclusive of mRNA-based interventions.
- Microbiology. From the moment the ability to map RNA modifications, specifically m6A, using an antibody-based sequencing technique became a reality, virologists have been keen to assess whether RNA produced from their virus of choice is also modified (Gokhale et al., 2016; Imam et al., 2018; Kennedy et al., 2016; McIntyre et al., 2018). These studies have provided great insights into previously unknown biology of viruses, such as by revealing that 2’Ome modifications of HIV-1 RNA can disguise the viral RNA from innate immune activators (Ringeard et al., 2019), or that ac4C can increase viral RNA stability (Tsai et al., 2020). Through the establishment of robust methodologies for the mapping of a variety of RNA modifications, these techniques will migrate over to molecular virology where researchers will be able to easily map the epitranscriptomes of viruses. These insights could readily provide some therapeutic potential. If modifications are found to be essential to viral replication, then antivirals can be designed specifically to block modification of viral RNA and thus block infection. Outside of virology, microbiologists would be strongly impacted by the work laid out here, as these technologies should also allow for the sequencing of the epitranscriptome of any bacteria or other unicellular organisms.
- Forensic science. The integration of epitranscriptomic analysis into forensic sciences could offer additional layers of information beyond traditional DNA profiling methods. Incorporating profiles from RNA modification may enhance forensic test sensitivity; enable multiplexing from crime scene samples (Sabeeha and Hasnain, 2019); and improve age estimation, tissue identification, and assessments of environmental exposures. However, the ethical considerations concerning the use of this epitranscriptomic information in legal and criminal contexts must be carefully addressed before any decisions on their use can be made.
- Big data, informatics and artificial intelligence. This project may push boundaries and generate an unprecedented amount of data, necessitating the development of better databases, larger data storage options, more advanced machine learning algorithms, and improved AI integration.
- Collaboration and open science. As happened with the HGP and the creation of atlases and databases such as those of the NCBI, the TCGA, etc., this project will once again prove the power of international collaboration and will highlight the importance and the advantage of open science. The data generated from this project will be deposited on the epigenetic atlas so researchers can share their data.
- Establishing a new state-of-the-art. As discussed above, this project will drive the advancement of current methodologies and inspire the creation of novel technologies and data analysis pipelines resulting in faster, more sensitive, and cost-effective approaches. The proposed collaborative effort will accelerate progress in the field, establish a baseline for sound and robust reporting of epitranscriptomic research, and ultimately lead to transformative discoveries and therapeutic breakthroughs.
Acknowledgements
David G. Courtney and his research are funded by a European Research Council starting grant, PTFLU 949506.
References
Anderson, A.C., I. Yanai, L. R. Yates, L. Wang, A. Swarbrick, P. Sorger, S. Santagata, W. H. Fridman, Q. Gao, L. Jerby, B. Izar, L. Shang, and X. Zhou. 2022. “Spatial transcriptomics.” Cancer Cell 40(9): 895–900. https://doi.org/10.1016/j.ccell.2022.08.021.
Arango, D., D. Sturgill, N. Alhusaini, A. A. Dillman, T. J. Sweet, G. Hanson, M. Hosogane, W. R. Sinclair, K. K. Nanan, M. D. Mandler, S. D. Fox, T. T. Zengeya, T. Andresson, J. L. Meier, J. Coller, and S. Oberdoerffer. 2018. “Acetylation of cytidine in mRNA promotes translation efficiency.” Cell 175(7): 1872–1886.E24. https://doi.org/10.1016/j.cell.2018.10.030.
Arzalluz-Luque, A, and A. Conesa. 2018. “Single-cell RNAseq for the study of isoforms—How is that possible?” Genome Biology 19(1):110. https://doi.org/10.1186/s13059-018-1496-z.
Baylin, S. B., and P. A. Jones. 2016. “Epigenetic determinants of cancer.” Cold Spring Harbor Perspectives in Biology 8(9):A019505. https://doi.org/10.1101/cshperspect.a019505.
Carlile, T. M., M. F. Rojas-Duran, B. Zinshteyn, H. Shin, K. M. Bartoli, and W. V. Gilbert. 2014. “Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells.” Nature 515 (7525): 143–146. https://doi.org/10.1038/nature13802.
Chen, H., J. Yao, R. Bao, Y. Dong, T. Zhang, Y. Du, G. Wang, D. Ni, Z. Xun, X. Niu, Y. Ye, and H-B. Li. 2021. “Crosstalk of four types of RNA modification writers defines tumor microenvironment and pharmacogenomic landscape in colorectal cancer. Molecular Cancer 20(1):29. https://doi.org/10.1186/s12943-021-01322-w.
Cook-Deegan, R.M. 1989. “The Alta Summit, December 1984.” Genomics 5(3):661–663. https://doi.org/10.1016/08887543(89)90042-6.
Courtney, D. G. 2021. “Post-transcriptional regulation of viral RNA through epitranscriptional modification.” Cells 10(5): 1129. https://doi.org/10.3390/cells10051129.
CSHL (Cold Springs Harbor Library). 1986. “Genome sequencing workshop (3-4 March 1986), 1986.” James D. Watson Collection. https://archivesspace.cshl.edu/repositories/2/archival_objects/65048 (accessed July 27, 2023).
Delehanty, J., R. L. White, and M. L. Mendelsohn. 1986. ICPEMC meeting report No. 2: Approaches to determining mutation rates in human DNA. Mutation Research/Reviews in Genetic Toxicology 167(3):215–232. https://doi.org/10.1016/0165-1110(86)90031-x.
Fleming, A. M., P. Bommisetti, S. Xiao, V. Bandarian, and C. J. Burrows. 2023. “Direct nanopore sequencing for the 17 RNA modification types in 36 locations in the E. coli ribosome enables monitoring of stress-dependent changes.” ACS Chemical Biology 18(10):2211–2223. https://doi.org/10.1021/acschembio.3c00166.
Garcia-Campos, M. A., S. Edelheit, U. Toth, M. Safra, R. Shachar, S. Viukov, R. Winkler, R. Nir, L. Lasman, A. Brandis, J. H. Hanna, W. Rossmanith, and S. Schwartz. 2019. “Deciphering the ‘m6A code’ via antibody-independent quantitative profiling.” Cell 178 (3): 731–747.E16. https://doi.org/10.1016/j.cell.2019.06.013.
Gibbs, R.A. 2020. “The human genome project changed everything.” Nature Reviews Genetics 21(10):575–576. https://doi.org/10.1038/s41576-020-0275-3.
Gokhale, N. S., A. B. R. McIntyre, M. J. McFadden, A. E. Roder, E. M. Kennedy, J. A. Gandara, S. E. Hopcraft, K. M. Quicke, C. Vazquez, J. Willer, O. R. Ilkayeva, B. A. Law, C. L. Holley, M. A. Garcia-Blanco, M. J. Evans, M. S. Suthar, S. S. Bradrick, C. E. Mason, and S. M. Horner. 2016. “N6-methyladenosine in flaviviridae viral RNA genomes regulates infection.” Cell Host & Microbe 20(5):654–665. https://doi.org/10.1016/j.chom.2016.09.015.
Hood, L., and L. Rowen. 2013. “The human genome project: Big science transforms biology and medicine.” Genome Medicine 5(9):79. https://doi.org/10.1186/gm483.
Hsu, P. J., Q. Fei, Q. Dai, H. Shi, D. Dominissini, L. Ma, and C. He. 2018. “Single base resolution mapping of 2'-O-meth-ylation sites in human MRNA and in 3’ terminal ends of small RNAs.” Methods 156: 85-90. https://doi.org/10.1016/j.ymeth.2018.11.007.
Hunter, M. V., R. Moncada, J. M. Weiss, I. Yanai, and R. M. White. 2021. “Spatially resolved transcriptomics reveals the architecture of the tumor-microenvironment interface.” Nature Communications 12(1):6278. https://doi.org/10.1038/s41467-021-26614-z.
Imam, H., M. Khan, N. S. Gokhale, A. B. R. McIntyre, G-W. Kim, J. Y. Jang, S-J. Kim, C. E. Mason, S. M. Horner, and A. Siddiqui. 2018. “N6 -methyladenosine modification of hepatitis B virus RNA differentially regulates the viral life cycle.” Proceedings of the National Academy of Sciences of the United States of America 115(35):8829–8834. https://doi.org/10.1073/pnas.1808319115.
Janin, M., V. Davalos, and M. Esteller. 2023. “Cancer metastasis under the magnifying glass of epigenetics and epitranscriptomics.” Cancer Metastasis Reviews 42(2):1071–1112. https://doi.org/10.1007/s10555-023-10120-3.
Karikó, K., M. Buckstein, H. Ni, and D. Weissman. 2005. “Suppression of RNA recognition by Toll-like receptors: The impact of nucleoside modification and the evolutionary origin of RNA.” Immunity 23 (2): 165–175. https://doi.org/10.1016/j.immuni.2005.06.008.
Kennedy, E. M., H. P. Bogerd, A. V. R. Kornepati, D. Kang, D. Ghoshal, J. B. Marshall, B. C. Poling, K. Tsai, N. S. Gokhale, S. M. Horner, and B. R. Cullen. 2016. “Posttranscriptional M(6)A editing of HIV-1 MRNAs enhances viral gene expression.” Cell Host & Microbe 19(5): 675–685. https://doi.org/10.1016/j.chom.2016.04.002.
Lander, E. S., L. M. Linton, B. Birren, C. Nusbaum, M. C. Zody, J. Baldwin, K. Devon, K. Dewar, M. Doyle, W. FitzHugh, R. Funke, D. Gage, K. Harris, A. Heaford, J. Howland, L. Kann, J. Lehoczky, R. LeVine, P. McEwan, K. McKernan, J. Meldrim, J. P. Mesirov, C. Miranda, W. Morris, J. Naylor, C. Raymond, M. Rosetti, R. Santos, A. Sheridan, C. Sougnez, Y. Stange-Thomann, N. Stojanovic, A. Subramanian, D. Wyman, J. Rogers, J. Sulston, R. Ainscough, S. Beck, D. Bentley, J. Burton, C. Clee, N. Carter, A. Coulson, R. Deadman, P. Deloukas, A. Dunham, I. Dunham, R. Durbin, L. French, D. Grafham, S. Gregory, T. Hubbard, S. Humphray, A. Hunt, M. Jones, C. Lloyd, A. McMurray, L. Matthews, S. Mercer, S. Milne, J. C. Mullikin, A. Mungall, R. Plumb, M. Ross, R. Shownkeen, S. Sims, R. H. Waterston, R. K. Wilson, L. W. Hillier, J. D. McPherson, M. A. Marra, E. R. Mardis, L. A. Fulton, A. T. Chinwalla, K. H. Pepin, W. R. Gish, S. L. Chissoe, M. C. Wendl, K. D. Delehaunty, T. L. Miner, A. Delehaunty, J. B. Kramer, L. L. Cook, R. S. Fulton, D. L. Johnson, P. J. Minx, S. W. Clifton, T. Hawkins, E. Branscomb, P. Predki, P. Richardson, S. Wenning, T. Slezak, N. Doggett, J. F. Cheng, A. Olsen, S. Lucas, C. Elkin, E. Uberbacher, M. Frazier, R. A. Gibbs, D. M. Muzny, S. E. Scherer, J. B. Bouck, E. J. Sodergren, K. C. Worley, C. M. Rives, J. H. Gorrell, M. L. Metzker, S. L. Naylor, R. S. Kucherlapati, D. L. Nelson, G. M. Weinstock, Y. Sakaki, A. Fujiyama, M. Hattori, T. Yada, A. Toyoda, T. Itoh, C. Kawagoe, H. Watanabe, Y. Totoki, T. Taylor, J. Weissenbach, R. Heilig, W. Saurin, F. Artiguenave, P. Brottier, T. Bruls, E. Pelletier, C. Robert, P. Wincker, D. R. Smith, L. Doucette-Stamm, M. Rubenfield, K. Weinstock, H. M. Lee, J. Dubois, A. Rosenthal, M. Platzer, G. Nyakatura, S. Taudien, A. Rump, H. Yang, J. Yu, J. Wang, G. Huang, J. Gu, L. Hood, L. Rowen, A. Madan, S. Qin, R. W. Davis, N. A. Federspiel, A. P. Abola, M. J. Proctor, R. M. Myers, J. Schmutz, M. Dickson, J. Grimwood, D. R. Cox, M. V. Olson, R. Kaul, C. Raymond, N. Shimizu, K. Kawasaki, S. Minoshima, G. A. Evans, M. Athanasiou, R. Schultz, B. A. Roe, F. Chen, H. Pan, J. Ramser, H. Lehrach, R. Reinhardt, W. R. McCombie, M. de la Bastide, N. Dedhia, H. Blöcker, K. Hornischer, G. Nordsiek, R. Agarwala, L. Aravind, J. A. Bailey, A. Bateman, S. Batzoglou, E. Birney, P. Bork, D. G. Brown, C. B. Burge, L. Cerutti, H. C. Chen, D. Church, M. Clamp, R. R. Copley, T. Doerks, S. R. Eddy, E. E. Eichler, T. S. Furey, J. Galagan, J. G. Gilbert, C. Harmon, Y. Hayashizaki, D. Haussler, H. Hermjakob, K. Hokamp, W. Jang, L. S. Johnson, T. A. Jones, S. Kasif, A. Kaspryzk, S. Kennedy, W. J. Kent, P. Kitts, E. V. Koonin, I. Korf, D. Kulp, D. Lancet, T. M. Lowe, A. McLysaght, T. Mikkelsen, J. V. Moran, N. Mulder, V. J. Pollara, C. P. Ponting, G. Schuler, J. Schultz, G. Slater, A. F. Smit, E. Stupka, J. Szustakowki, D. Thierry-Mieg, J. Thierry-Mieg, L. Wagner, J. Wallis, R. Wheeler, A. Williams, Y. I. Wolf, K. H. Wolfe, S. P. Yang, R. F. Yeh, F. Collins, M. S. Guyer, J. Peterson, A. Felsenfeld, K. A. Wetterstrand, A. Patrinos, M. J. Morgan, P. de Jong, J. J. Catanese, K. Osoegawa, H. Shizuya, S. Choi, Y. J. Chen, and J. Szustakowki. 2001. “Initial sequencing and analysis of the human genome.” Nature 409 (6822): 860–921. https://doi.org/10.1038/35057062.
Lebrigand, K., V. Magnone, P. Barbry, and R. Waldmann. 2020. “High throughput error corrected Nanopore single cell transcriptome sequencing.” Nature Communications 11(1):4025. https://doi.org/10.1038/s41467-020-17800-6.
Liu, E. T. 2009. “The human genome organisation (HUGO).” The Hugo Journal 3(1–4):3–4. https://doi.org/10.1007/s11568-010-9139-9.
Liu, W., C. Liu, H. Wang, L. Xu, J. Zhou, S. Li, Y. Cheng, R. Zhou, and L. Zhao. 2022. “Targeting N6-methyladenosine RNA modification combined with immune checkpoint inhibitors: A new approach for cancer therapy.” Computational and Structural Biotechnology Journal 20:5150-5161. https://doi.org/10.1016/j.csbj.2022.09.017.
Liu, W., Z-Y. Zhang, F. Wang, and H. Wang, H. 2023. “Emerging roles of m6a RNA modification in cancer therapeutic resistance.” Experimental Hematology & Oncology 12(1):21. doi:10.1186/S40164-023-00386-2.
Lucas, M. C., and E. M. Novoa. 2023. “Long-read sequencing in the era of epigenomics and epitranscriptomics.” Nature Methods 20(1): 25–29. https://doi.org/10.1038/s41592-022-01724-8.
Marchand, V., L. Ayadi, F. G. M. Ernst, J. Hertler, V. Bourguignon-Igel, A. Galvanin, A. Kotter, M. Helm, D. L. J. Lafontaine, and Y. Motorin. 2018. “AlkAniline-Seq: Profiling of m7g and m3c RNA modifications at single nucleotide resolution.” Angewandte Chemie 57(51):16785–16790. https://doi.org/10.1002/anie.201810946.
Marchand, V., F. Blanloeil-Oillo, M. Helm, and Y. Motorin. 2016. “Illumina-based RiboMethSeq approach for mapping of 2’-O-Me residues in RNA.” Nucleic Acids Research 44 (16): E135. https://doi.org/10.1093/nar/gkw547.
McIntyre, W., R. Netzband, G. Bonenfant, J. M. Biegel, C. Miller, G. Fuchs, E. Henderson, M. Arra, M. Canki, D. Fabris, and C. T. Pager. 2018. “Positive-sense RNA viruses reveal the complexity and dynamics of the cellular and viral epitranscriptomes during infection.” Nucleic Acids Research 46(11): 5776–5791. https://doi.org/10.1093/nar/gky029.
Miano, V., A. Codino, L. Pandolfini, and I. Barbieri. “The non-coding epitranscriptome in cancer.” Briefings in Functional Genomics 20(2):94-105. https://doi.org/10.1093/bfgp/elab003.
Morais, P., H. Adachi, and Y. T. Yu. 2021. “The critical contribution of pseudouridine to mRNA COVID-19 vaccines.” Frontiers in Cell and Developmental Biology 9: 789427. https://doi.org/10.3389/fcell.2021.789427.
Nance, K. D., and J. L. Meier. 2021. “Modifications in an emergency: The role of N1-methylpseudouridine in COVID-19 vaccines.” ACS Central Science 7 (5): 748–756. https://doi.org/10.1021/acscentsci.1c00197.
Netzband, R., and C. T. Pager. 2020. “Epitranscriptomic marks: Emerging modulators of RNA virus gene expression.” Wiley Interdisciplinary Reviews: RNA 11(3):E1576. https://doi.org/10.1002/wrna.1576.
Rasool, M., A. Malik, M. I. Naseer, A. Manan, S. Ansari, I. Begum, M. H. Qazi, P. Pushparaj, A. M. Abuzenadah, M. H. Al-Qahtani, M. A. Kamal, and S. Gan. 2015. “The role of epigenetics in personalized medicine: Challenges and opportunities.” BMC Medical Genomics 8(Suppl 1):S5. https://doi.org/10.1186/1755-8794-8-s1-s5.
Ringeard, M., V. Marchand, E. Decroly, Y. Motorin, and Y. Bennasser. 2019. “FTSJ3 is an RNA 2′-O-methyltrans-ferase recruited by HIV to avoid innate immune sensing.” Nature 565(7740): 500–504. https://doi.org/10.1038/s41586-018-0841-4.
Sabeeha, and S. E. Hasnain. 2019. “Forensic epigenetic analysis: The path ahead.” Medical Principles and Practice 28(4): 301–308. https://doi.org/10.1159/000499496.
Sas-Chen, A., J. M. Thomas, D. Matzov, M. Taoka, K. D. Nance, R. Nir, K. M. Bryson, R. Shachar, G. L. S. Liman, B. W. Burkhart, S. T. Gamage, Y. Nobe, C. A. Briney, M. J. Levy, R. T. Fuchs, G. B. Robb, J. Hartmann, S. Sharma, Q. Lin, L. Florens, M. P. Washburn, T. Isobe, T. J. Santangelo, M. Shalev-Benami, J. L. Meier, and S. Schwartz. 2020. “Dynamic RNA acetylation revealed by quantitative cross-evolutionary mapping.” Nature 583 (7817): 638–643. https://doi.org/10.1038/s41586-020-2418-2.
Schaefer, M., T. Pollex, K. Hanna, and F. Lyko. 2009. “RNA cytosine methylation analysis by bisulfite sequencing.” Nucleic Acids Research 37 (2): E12. https://doi.org/10.1093/nar/gkn954.
Schwartz, S., D. A. Bernstein, M. R. Mumbach, M. Jovanovic, R. H. Herbst, B. X. León-Ricardo, J. M. Engreitz, M. Gutt-man, R. Satija, E. S. Lander, G. Fink, and A. Regev. 2014. “Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA.” Cell 159 (1): 148–162. https://doi.org/10.1016/j.cell.2014.08.028.
Sturgill, D., D. Arango, and S. Oberdoerffer. 2022. “Protocol for base resolution mapping of ac4c using RedaC:T-Seq.” STAR Protocols 3(4):101858. https://doi.org/10.1016/j.xpro.2022.101858.
Sun, H., Y. Zhang, G. Wang, W. Yang, and Y. Xu. 2023. “MRNA-based therapeutics in cancer treatment.” Pharmaceutics 15(2):622. https://doi.org/10.3390/pharmaceutics15020622.
Thalalla Gamage, S., A. Sas-Chen, S. Schwartz, and J. L. Meier. 2021. “Quantitative nucleotide resolution profiling of RNA cytidine acetylation by ac4C-seq.” Nature Protocols 16(4):2286-2307. https://doi.org/10.1038/s41596-021-00501-9.
Tsai, K., and B. R. Cullen. 2020. Epigenetic and epitranscriptomic regulation of viral replication. Nature Reviews Microbiology 18(10): 559–570. https://doi.org/10.1038/s41579-020-0382-3.
Tsai, K., A. A. J. Vasudevan, C. Martinez Campos, A. Emery, R. Swanstrom, and B. R. Cullen. 2020. “Acetylation of cytidine residues boosts HIV-1 gene expression by increasing viral RNA stability.” Cell Host & Microbe 28(2):306–312. https://doi.org/10.1016/j.chom.2020.05.011.
Venter, J. C., M. D. Adams, E. W. Myers, P. W. Li, R. J. Mural, G. G. Sutton, H. O. Smith, M. Yandell, C. A. Evans, R. A. Holt, J. D. Gocayne, P. Amanatides, R. M. Ballew, D. H. Huson, J. R. Wortman, Q. Zhang, C. D. Kodira, X. H. Zheng, L. Chen, M. Skupski, G. Subramanian, P. D. Thomas, J. Zhang, G. L. Gabor Miklos, C. Nelson, S. Broder, A. G. Clark, J. Nadeau, V. A. McKusick, N. Zinder, A. J. Levine, R. J. Roberts, M. Simon, C. Slayman, M. Hunkapiller, R. Bolanos, A. Delcher, I. Dew, D. Fasulo, M. Flanigan, L. Florea, A. Halpern, S. Hannenhalli, S. Kravitz, S. Levy, C. Mobarry, K. Reinert, K. Remington, J. Abu-Threideh, E. Beasley, K. Biddick, V. Bonazzi, R. Brandon, M. Cargill, I. Chandramouliswaran, R. Charlab, K. Chaturvedi, Z. Deng, V. Di Francesco, P. Dunn, K. Eilbeck, C. Evangelista, A. E. Gabrielian, W. Gan, W. Ge, F. Gong, Z. Gu, P. Guan, T. J. Heiman, M. E. Higgins, R. R. Ji, Z. Ke, K. A. Ketchum, Z. Lai, Y. Lei, Z. Li, J. Li, Y. Liang, X. Lin, F. Lu, G. V. Merkulov, N. Milshina, H. M. Moore, A. K. Naik, V. A. Narayan, B. Neelam, D. Nusskern, D. B. Rusch, S. Salzberg, W. Shao, B. Shue, J. Sun, Z. Wang, A. Wang, X. Wang, J. Wang, M.-H. Wei, R. Wides, C. Xiao, C. Yan, A. Yao, J. Ye, M. Zhan, W. Zhang, H. Zhang, Q. Zhao, L. Zheng, F. Zhong, W. Zhong, S. Zhu, S. Zhao, D. Gilbert, S. Baumhueter, G. Spier, C. Carter, A. Cravchik, T. Woodage, F. Ali, H. An, A. Awe, D. Baldwin, H. Baden, M. Barnstead, I. Barrow, K. Beeson, D. Busam, A. Carver, A. Center, M. L. Cheng, L. Curry, S. Danaher, L. Davenport, R. Desilets, S. Dietz, K. Dodson, L. Doup, S. Ferriera, N. Garg, A. Gluecksmann, B. Hart, J. Haynes, C. Haynes, C. Heiner, S. Hladun, D. Hostin, J. Houck, T. Howland, C. Ibegwam, J. Johnson, F. Kalush, L. Kline, S. Koduru, A. Love, F. Mann, D. May, S. McCawley, T. McIntosh, I. McMullen, M. Moy, L. Moy, B. Murphy, K. Nelson, C. Pfannkoch, E. Pratts, V. Puri, H. Qureshi, M. Reardon, R. Rodriguez, Yu-Hui Rogers, D. Romblad, B. Ruhfel, R. Scott, C. Sitter, M. Smallwood, E. Stewart, R. Strong, E. Suh, R. Thomas, N. N. Tint, S. Tse, C. Vech, G. Wang, J. Wetter, S. Williams, M. Williams, S. Windsor, E. Winn-Deen, K. Wolfe, J. Zaveri, K. Zaveri, J. F. Abril, R. Guigó, M. J. Campbell, K. V. Sjolander, B. Karlak, A. Kejariwal, H. Mi, B. Lazareva, T. Hatton, A. Narechania, K. Diemer, A. Muruganujan, N. Guo, S. Sato, V. Bafna, S. Istrail, R. Lippert, R. Schwartz, B. Walenz, S. Yooseph, D. Allen, A. Basu, J. Baxendale, L. Blick, M. Caminha, J. Carnes-Stine, P. Caulk, Yen-Hui Chiang, M. Coyne, C. Dahlke, A. Deslattes Mays, M. Dombroski, M. Donnelly, D. Ely, S. Esparham, C. Fosler, H. Gire, S. Glanowski, K. Glasser, A. Glodek, M. Gorokhov, K. Graham, B. Gropman, M. Harris, J. Heil, S. Henderson, J. Hoover, D. Jennings, C. Jordan, J. Jordan, J. Kasha, L. Kagan, C. Kraft, A. Levitsky, M. Lewis, X. Liu, J. Lopez, D. Ma, W. Majoros, J. McDaniel, S. Murphy, M. Newman, T. Nguyen, N. Nguyen, M. Nodell, S. Pan, J. Peck, M. Peterson, W. Rowe, R. Sanders, J. Scott, M. Simpson, T. Smith, A. Sprague, T. Stockwell, R. Turner, E. Venter, M. Wang, M. Wen, D. Wu, M. Wu, A. Xia, A. Zandieh, and X. Zhu. 2001. “The sequence of the human genome.” Science 291 (5507): 1304–1351. https://doi.org/10.1126/science.1058040.
Yang, D., G. Zhao, and H. M. Zhang. 2023. “m6a reader proteins: The executive factors in modulating viral replication and host immune response.” Frontiers in Cellular and Infection Microbiology 13. https://doi.org/10.3389/fcimb.2023.1151069.
Zhang, L-S., C. Liu, H. Ma, Q. Dai, H-L. Sun, G. Luo, Z. Zhang, L. Zhang, L. Hu, Z. Dong, and C. He. 2019. “Transcrip-tome-wide mapping of internal N7-methylguanosine methylome in mammalian mRNA.” Molecular Cell 74(6):1304–1316.E8. https://doi.org/10.1016/j.molcel.2019.03.036.
Zhang, L-S., Q. Dai, and C. He. 2023. “BID-Seq: The quantitative and base-resolution sequencing method for RNA pseudouridine.” ACS Chemical Biology 18(1): 4–6. https://doi.org/10.1021/acschembio.2c00881.
Zhang, M., Z. Jiang, Y. Ma, W. Liu, Y. Zhuang, B. Lu, K. Li, J. Peng, and C. Yi. 2023. “Quantitative profiling of pseudouridylation landscape in the human transcriptome.” Nature Chemical Biology 19(10):1185–1195. https://doi.org/10.1038/s41589-023-01304-7.
Zhang, Y., L-S. Zhang, Q. Dai, P. Chen, M. Lu, E. L. Kairis, V. Murugaiah, J. Xu, R. K. Shukla, X. Liang, Z. Zou, E. Cormet-Boyaka, J. Qiu, M. E. Peeples, A. Sharma, C. He, and J. Li. 2022. “5-methylcytosine (m5c) RNA modification controls the innate immune response to virus infection by regulating type I interferons.” Proceedings of the National Academy of Sciences of the United States of America 119(42):E2123338119. https://doi.org/10.1073/pnas.2123338119.
Zhou, H., S. Rauch, Q. Dai, X. Cui, Z. Zhang, S. Nachtergaele, C. Sepich, C. He, and B. C. Dickinson. 2019. “Evolution of a reverse transcriptase to map N1-methyladenosine in human messenger RNA.” Nature Methods 16(12): 1281–1288. https://doi.org/10.1038/s41592-019-0550-4.















































