Firepower in the Lab: Automation in the Fight Against Infectious Diseases and Bioterrorism (2001)
Chapter: Flow Cytometry Analysis Techniques for High-Throughput Biodefense Research
17
Flow Cytometry Analysis Techniques for High-Throughput Biodefense Research
James H. Jett, Hong Cai, Robert C. Habbersett, Richard A. Keller, Erica J. Larson, Babetta L. Marrone, John P. Nolan, Xuedong Song, Basil Swanson, and Paul S. White
INTRODUCTION
Contemporary flow cytometry has its roots in developments of rapid cellular analyses that began in the 1960s. Initial measurements focused on the analysis of hematopoietic and other mammalian cell types to determine size distributions, DNA content distributions, and surface antigen expression distributions (Melamed et al., 1990). With the advent of monoclonal antibodies, studies of immune systems by flow cytometry exploded, and important fundamental aspects are still being discovered. Over the years, the application of flow cytometry has moved from the analysis of whole cells and their constituents to that of subcellular components. Through improvements in detection sensitivity, it is now possible to analyze individual molecules (Ambrose et al., 1999). Thus, the name “cytometer,” or cell analyzer, is not appropriate today. A more descriptive name for the technology is “fluorescent particle analysis in a flowing sample stream.” Several applications of the basic technology to high-throughput research are emerging in the biodefense arena.
The key to understanding the capabilities of flow cytometric measurements is contained in the method of sample presentation. In a generic sense, objects in suspension to be analyzed are introduced into the measurement region after hydrodynamic focusing by a surrounding sheath fluid. This is depicted schematically in Figure 17.1. In addition to confining the sample stream to a very narrow diameter, typically 10 microns, the sample velocity is increased. The fully focused sample stream passes through one or more focused laser beams. In addition to scattering light,

FIGURE 17.1 A schematic drawing of a typical flow cytometer illustrating the single-particle sample inlet tube at the top and the hydrodynamic focusing of the sample stream as it is transported through one or more laser beams. Fluorescence is collected at right angles to the laser beam sample stream axes, filtered optically, and focused onto detectors.
fluorescent dyes bound to specific components of the objects being analyzed are excited and emit fluorescence photons. The laser beam transit time is roughly dependent on the measurement sensitivity required. For cellular analyses, the transit time is typically 10 microseconds. This translates into analysis rates ranging up to tens of thousands of particles per second. For single-molecule detection and DNA fragment size analysis, the transit times are measured in milliseconds, which limits the analysis rates to roughly 100 molecules per second.
The fluorescence photons emitted as the particles pass through the laser beam are collected, filtered spectrally, and directed to one or more
detectors. Detector signals are processed to derive up to three types of information: transit time through the laser beam, maximum fluorescence emission, and total fluorescence emission. These pieces of information from each detector are correlated with other measurements, such as those listed below, for the particle and recorded in the computer. Subsequent offline analysis of the multidimensional data collected is often required to extract the biological information of interest.
For the higher-velocity cell analysis systems, it is possible to physically separate (sort) the particles by forcing the stream exiting the flow cell to form droplets at an induced frequency. The optimal frequency is roughly inversely proportional to the exit orifice diameter, ranging from 5 kHz for a 200-micron-diameter orifice to 70 kHz for a 50-micron-orifice. Measurements made as the particles transit the laser beam(s) are used to define which particles are to be separated from the parent population. When a particle whose measurements satisfy the sort criteria is contained in the terminal droplet that is ready to break off from the solid stream above, a charge is applied to the stream. Once the droplet has broken off, the charge is removed from the stream, but the droplet remains charged as it falls between deflection plates that are held at a constant high voltage. The deflected droplets are collected in a vessel, while the particles not of interest are not deflected.
In addition to measuring the total or maximum fluorescence emitted in multiwavelength regions, other properties of the fluorescence emissions have been measured, including polarization state and lifetime. Scattered-light measurements, when properly analyzed, yield information about a particle's size and surface characteristics.
Two examples of flow cytometric measurements that have application in the biodefense arena are DNA fragment size analysis and microsphere-based assays for multiplexed minisequencing and toxin detection. Currently, the first two of these technologies are being developed to identify bacteria, bacterial strains, and bacterial pathogenicity.
DNA FRAGMENT SIZING FLOW CYTOMETRY
DNA fragment size distribution analysis is a ubiquitous measurement in molecular biology. Applications exist in disease diagnostics, large insert DNA clone analysis, bacteria strain and species identification, polymerase chain reaction (PCR) product analysis, and forensics. Based on our experience in individual fluorescent analysis (Ambrose et al., 1999), we have developed a new technique for sizing DNA fragments based on flow cytometry (Goodwin et al., 1993; Petty et al., 1995; Habbersett et al., 2000). For large DNA fragments—fragments greater than 30 kilobase pairs—our approach is more sensitive (sample size of femtograms versus
micrograms), faster (analysis time of 5 minutes versus tens of hours), and more accurate (size uncertainty of 2 percent versus 10 percent) than pulse-field gel electrophoresis (PFGE), commonly used for these analyses.
State-of-the-art techniques for DNA fragment size analysis use some form of electrophoresis. For samples containing DNA fragments greater than 30 kilobase pairs in length, PFGE methods are used that require up to 24 hours or more to achieve the desired separations of hundreds of nanograms of DNA. Furthermore, all forms of electrophoretic separation are highly nonlinear, which affects the accuracy of fragment size measurements.
PFGE is often used in hospital settings and in state public health laboratories to determine the presence of an outbreak or to make a diagnosis of a bacterial infection. Currently, sample preparation methods are carried out in a plug of the gel matrix in which the bacterial sample is embedded and processed. Due to long diffusion times of reagents into and out of the plug, the process can require up to 7 days to complete before the PFGE analysis. Recent developments at Los Alamos and elsewhere (Chang and Chui, 1998) have reduced the sample preparation time to 8 hours or less.
In general, a bacterial sample is embedded in an agarose matrix and reagents are diffused into and out of the agarose that break down the bacterial cell wall, digest the proteins, and cut up the bacterial genome with restriction enzymes. The restriction endonuclease severs the bacterial DNA at specific sequence sites. Typically, for bacterial species and strain identification, the restriction sites are eight bases long, which results in an average fragment length of 65 kilobase pairs. Eight-base instead of more frequent cutters are used to keep the number of fragments produced per genome manageable. Analysis of the distribution of fragment sizes by PFGE or flow cytometry produces a “fingerprint” pattern that is specific to the bacterial species and strain being analyzed. Often, the restriction digest fingerprint can uniquely identify the organism.
For analysis of bacterial genome restriction digests by flow cytometry, the DNA sample, which is prepared in the same manner as for PFGE analysis, is stained with a fluorescent intercalating dye that binds stoichio-metrically to the DNA such that the amount of dye incorporated is directly proportional to the fragment size (number of base pairs [bp]). The 1,000-fold increase in the fluorescence quantum yield of the intercalating dye upon binding to the DNA makes it unnecessary to remove unbound dye from the solution before analysis. The stained fragments are diluted to 10−12 to 10−14 molar and introduced into an ultrasensitive flow cytometer. Fragments pass individually through the laser-illuminated detection region of the flow cytometer, each fragment producing a burst of fluorescence photons as it transits the laser beam. Fluorescence bursts from individual fragments are quantified and recorded. The burst size is a measure
of the fragment size. A histogram of the burst sizes is generated that displays the distribution of fragment sizes in the sample (i.e., a DNA fingerprint). The response linearity has been verified stepwise over the size range analyzed to date: 125 to 450,000 bp.
Histograms of the burst sizes resulting from the analysis of restriction digests of four Staphylococcus aureus strains are shown in Figure 17.2. Each histogram was compiled from less than 5 minutes of data (15,000 events) acquired from less than a femtogram of DNA. Plots of the burst sizes

FIGURE 17.2 Examples of the restriction digest fingerprint patterns by four strains of S. aureus. The inserts show the relationship between the fragment sizes measured by flow cytometry and the sizes determined by PFGE.
versus the fragment lengths determined by PFGE analyses also are displayed. The plots are linear with correlation coefficients of r2 > 0.999. Most of the deviation of the points from the fit line is attributed to the 10 percent uncertainty inherent in PFGE versus the 2 percent uncertainty of flow cytometric measurements. Each strain produced a distinct fingerprint. Similar results have been obtained for several strains of Escherichia coli (Larson et al., 2000).
Application of this technology to bacterial species and strain identification is being pursed vigorously. Macrorestriction digests of bacterial genomes with infrequently cutting restriction endonucleases analyzed by PFGE provide one of the most definitive signatures for species and strain identification (Maslow et al., 1993a, 1993b). These analyses typically require micrograms of DNA, 8 hours for sample processing, followed by ~20 hours for PFGE separation and analysis of the results. The patterns observed, which vary with restriction enzyme used as well as with species and strain analyzed, form the basis for identifying the bacterial species and strain.
A description of species discrimination by flow cytometric DNA fragment sizing has been published (Kim et al., 1999). Figure 17.2 demonstrates an example of flow cytometric measurements of restriction digests of four strains of S. aureus digested with the restriction enzyme Sma I. In addition to sample preparation time being reduced to 6 hours followed by 5 minutes of analysis, the amount of DNA required is one-millionth the amount needed for PFGE analysis.
There are several specific biodefense applications of DNA fragment sizing technology. The technology is capable of recognizing and identifying both bacterial species and strains. This capability is useful in forensic applications, where attribution to a source of a biothreat agent used in an incident is desired, in public health laboratories for identifying and tracking outbreaks, and at special events to confirm the identity of biothreat agents by other means. For nonproliferation assays it will be possible to develop profiles of the effluents from production facilities and to confirm organism identification in treaty verification studies. Highly automated throughput sample preparation and analysis systems will be developed in the future to enable the processing and analysis of large numbers of samples.
FLOW CYTOMETRIC MICROSPHERE-BASED ASSAYS
A characteristic of a conventional flow cytometer is that, since the sample is confined to move in a narrow stream and the excitation laser is tightly focused, the volume that is observed by the detection optics can be small (pL – nL), and fluorescence emissions originating outside the probe
volume are not “seen” by the detector. The operational result of this characteristic is that it is not necessary to separate particle-bound from free fluorescent molecules, which enables the performance of a wide variety of homogeneous assays.
Microspheres in the 3- to 10-micron diameter range are used to carry the molecular assemblies for a number of biomolecular assays that use a flow cytometer as a readout instrument. Applications of microsphere-based assays include measurement of enzyme activity (Frank et al., 1998), cholera toxin detection (Song et al., 1998), immunoassays (Saunders et al., 1985), and single nucleotide polymorphism (SNP) detection (Cai et al., 2000). SNP detection is an example of the application of minisequencing where the reagents are carried on a microsphere.
In Figure 17.3 the circle represents a microsphere to which is attached a large number (~105) of oligonucleotides that end one position short of the site of interest. DNA from the sample (beginning and ending with. ) being analyzed is hybridized to the oligonucleotides attached to the microsphere. The “C” is added to the top oligo by enzymatic incorporation of a dideoxy nucleotide that is fluorescently labeled. This reaction is carried out in four separate tubes, each containing a different labeled nucleotide, along with three unlabeled dideoxy nucleotides. Thus, when each of the four samples is analyzed, only one sample will be measured as having fluorescence bound to the microsphere—the one with the correctly labeled nucleotide incorporated. As biothreat organisms are sequenced, this technique will be used to determine agent species and strains.
The microsphere-based cholera toxin assay uses a different principle, that of fluorescence resonant energy transfer. When cholera toxin binds to a cell, multiple receptor molecules bind to the toxin. In this assay the microspheres are coated with a lipid bilayer in which are embedded two types of cholera toxin receptor molecules. The receptor molecules are labeled with either a fluorescence donor or acceptor. In the absence of the toxin, the donor-acceptor-labeled molecules are far enough apart that, when the donor molecule is excited by an appropriate wavelength of laser light, fluorescence energy transfer does not occur, resulting in a fluorescence emission spectrum from the bead that is characteristic of the donor molecule. However, when toxin is present, donor acceptor pairs
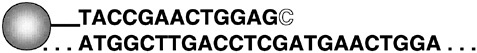
FIGURE 17.3 Schematic representation of the minisequencing process.
are brought into close proximity (~5 nm), and the fluorescence emission spectrum becomes more characteristic of the acceptor molecule. By spectral selection the ratio of the donor to acceptor fluorescence can be measured and, once calibrated, provides a measure of the cholera toxin concentration (Song et al., 1998).
FUTURE DIRECTIONS
To this point, single assays that are bead based have been discussed. A new technology has been developed that enables multiplexing of assays. A bead set developed by the Luminex Corporation (Austin, Tex.; www.luminexcorp.com) consists of beads that are stained with two dyes in varying amounts. The bead types are distinguishable by measuring the amount of fluorescence in two wavelength regions. The assay results, such as the minisequencing assay, are reported in a third wavelength region. Thus, a flow cytometer capable of recording fluorescence emissions in three wavelength regions is able to separate the bead types and record the assay results reported by each type. Currently, bead sets containing 64 bead types are available, with larger sets planned for the future. In addition to distinguishing beads by the amount of labeling dyes contained in them, it is possible to envision adding size, and perhaps shape, to provide other dimensions for multiplexing.
For high-throughput flow cytometric assays, automated instrument operation including sample introduction will be necessary. A recent development (Edwards et al., 1999) in sample introduction has achieved a sample throughput rate of nine samples per minute with less than 4 percent overlap between samples. The approach used is basically to line samples up in the sample delivery tube of a cytometer separated by regions of fluid that contain no particles. A series of computer-controlled valves coupled to a syringe drive are used to deliver the samples continuously to the cytometer. This and other developments in the future will enable truly high-throughput sample preparation and analysis that will take advantage of the flow cytometer's ability to analyze large numbers of individual particles in a sample at rates of thousands per second.
ACKNOWLEDGMENTS
This work was supported by the U.S. Department of Energy (NN-20), the National Institutes of Health National Center for Research Resources (Grant RR-01315), and the Los Alamos National Laboratory Laboratory Directed Research and Development program.
REFERENCES
Ambrose, W. P., P. M. Goodwin, J. H. Jett, A. Van Orden, J. H. Werner, and R. A. Keller. 1999. Single molecule fluorescence spectroscopy at ambient temperature. Chemical Reviews, 99:2929-2956.
Cai, H., P. S. White, D. C. Torney, A. Deshpande, Z. Wang, B. Marrone, and J. P. Nolan. 2000. Flow cytometry-based minisequencing: A new platform for high-throughput single-nucleotide polymorphism scoring. Genomics, 66:135-143.
Chang, N., and L. Chui. 1998. A standardized protocol for the rapid preparation of bacterial DNA for pulsed-field gel electrophoresis. Diagnostic Microbiology and Infectious Disease, 31:275-279.
Edwards, B. S., F. Kuckuck, and L. A. Sklar. 1999. Plug flow cytometry: An automated coupling device for rapid sequential flow cytometric sample analysis. Cytometry, 37:156-169.
Frank, Q., M. Somsouk, Y. Weng, L. Somsouk, J. P. Nolan, and B. Shen. 1998. Partial functional deficiency of E160D flap endonuclease-1 mutant in vitro and in vivo is due to defective cleavage of DNA substrates Journal of Biological Chemistry, 273:33064-33072.
Goodwin, P. M., M. E. Johnson, J. C. Martin, W. P. Ambrose, B. L. Marrone, J. H. Jett, and R. A. Keller. 1993. Rapid sizing of individual fluorescently stained DNA fragments by flow cytometry. Nucleic Acids Research, 21:803-806.
Habbersett, R. C., J. H. Jett, and R. A. Keller. 2000. Single fragment detection by flow cytometry. Pp. 115-138 in Emerging Tools for Cell Analysis: Advances in Optical Measurement J. P. Robinson and G. Durak, eds. New York: Wiley-Liss.
Kim, Y., J. H. Jett, E. J. Larson, J. R. Pentilla, B. L. Marone, and R. A. Keller. 1999. Bacterial fingerprinting by flow cytometry: Bacterial species discrimination Cytometry, 36:324-332.
Larson, E., J. R. Penttila, H. Cai, J. H. Jett, S. Burde, and R. A. Keller. 2000. Rapid DNA fingerprinting of pathogens by flow cytometry. Cytometry, 41:203-208.
Maslow, J. N., M. E. Mulligan, and R. D. Arbeit. 1993a. Molecular epidemiology: Application of contemporary techniques to the typing of microorganisms. Clinical Infectious Diseases, 17:153-164.
Maslow, J. N., A. M. Slutsky, and R. D. Arbeit. 1993b. Application of pulsed-field gel electrophoresis to molecular epidemiology In Diagnostic Molecular Microbiology: Principles and Applications, D. H. Persing, T. F. Smith, F. C. Tenover, and T. J. White, eds. Washington, D.C.: American Society for Microbiology.
Melamed, M. R., T. Lindmo, and M. L. Mendelsohn, eds. 1990. Flow Cytometry and Sorting. New York: Wiley-Liss.
Petty, J. T., M. E. Johnson, P. M. Goodwin, J. C. Martin, J. H. Jett, and R. A. Keller. 1995. Characterization of DNA size determination of small fragments by flow cytometry. Analytical Chemistry, 67:1755-1761.
Saunders, G. C., J. H. Jett, and J. C. Martin. 1985. Amplified flow cytometric separation free fluorescence immunoassay Clinical Chemistry, 31:2020-2023.
Song, X., J. P. Nolan, and B. Swanson. 1998. An optical biosensor based on fluorescence self-quenching and energy transfer: Ultrasensitive and specific detection of protein toxins Journal of the American Chemical Society, 120:11514-11515.