Carving Our Destiny: Scientific Research Faces a New Millennium (2001)
Chapter: Human Genetics in the Twenty-First Century
9
Human Genetics in the Twenty-First Century
Leonid Kruglyak
Division of Human Biology, Fred Hutchinson Cancer Research Center, Seattle, Washington
ABSTRACT
As human genetics enters the twenty-first century, many powerful molecular tools are either in place or about to arrive. They include a large capacity for DNA sequencing, an ability to examine many polymorphisms in many people, and techniques for monitoring expression levels of many genes under many conditions. These tools are already generating large amounts of data. Early in this century, we can envision obtaining complete DNA sequences of many individuals, expression levels of all genes in a variety of cells, and a complete characterization of protein levels and interactions. The key task facing human genetics is interpreting this data.
My research centers on addressing different aspects of this task. These include understanding the genetic basis of diseases and other phenotypes, the evolutionary history of human populations, and the regulatory networks of gene expression.
Genetic Basis of Disease
Genetic factors are thought to play some role in susceptibility to almost all diseases. We can search for these factors using several approaches. The most well-known entails tracing inheritance patterns of disease in families in order to implicate particular chromosomal regions
in disease transmission. Such studies have produced many important advances in our understanding of the genetic basis of diseases and other phenotypes, particularly those with simple Mendelian inheritance. For more common diseases, such as diabetes, heart disease, cancer, multiple sclerosis, asthma, manic depression, and schizophrenia, successes have been harder to come by because of the problems posed by genetic complexity. The genetic basis of such diseases is likely to consist of a large number of subtle effects, whose detection may require a prohibitively large number of families. Population studies offer a potential way to overcome this hurdle. The most direct approach is to start with a mutation and examine its effect on frequency of disease in populations. Alternatively, we can recognize that all people share ancestry sufficiently far back in time, and that many mutations in today’s population are likely to have a common origin. We can therefore search the DNA of population members for ancestral signatures of this origin.
Population History
The best repository of historical information is the DNA of population members. Patterns of spelling changes in the genetic code contain a record of population divergences, migrations, bottlenecks, replacements, and expansions. In addition to its inherent interest, this information provides important insights into the use of populations for studies of the genetic basis of disease, including the specific advantages offered by different populations. Molecular approaches have already contributed a great deal to our understanding of human evolution. These approaches need to be updated to take full advantage of modern molecular tools, provide a more detailed look at the entire genome, and resolve finer points of population diversity and relationships.
Regulatory Networks
The genetic code is a static record of information. The dynamic response of a cell to changing conditions is governed in large part by which genes are expressed—transcribed into messenger RNA. Several emerging technologies now allow simultaneous measurements of transcription levels of many genes in a cell under a variety of conditions. A key task is to reconstruct the network of interactions among genes from these observations. Understanding such regulatory networks is an important part of studying any biological process. Going beyond gene expression, many important dynamic responses involve modifications and interactions of proteins. Once technology allows proteins to be monitored with ease,
ideas developed to understand networks of interacting genes can be applied to this next level of complexity.
Genetic Research Must Be Applied Responsibly
A key aspect of this goal is education. We must make our research and its implications clearly and broadly understood. Armed with appropriate information, society will be prepared to face the complex questions posed by progress in human genetics, reaping the benefits while avoiding the pitfalls.
Genetic approaches extend beyond human genetics to studies of pathogens and organisms of agricultural importance, and even more broadly to a better understanding of genetics and biology of all organisms. The ultimate benefits of genetic research to society take many forms: better medicine, improved quality of life, increased knowledge, and a greater understanding of who we are and where we fit in among other forms of life.
INTRODUCTION
The second half of the twentieth century witnessed the development of molecular biology and its tremendous impact on our understanding of life. In the past two decades, molecular techniques have been applied to our own species, giving rise to human molecular genetics and the resulting revolution in medicine. The Human Genome Project is poised to generate a complete readout of the human genetic code by the year 2003. It is not too early to ask: How much of the secret of human life is contained in the complete DNA sequence of every person on the planet, and how can we extract it? My research centers on answering this question in its many forms, a task that will occupy human geneticists for much of the twenty-first century.
This scenario of obtaining the complete DNA sequence of every person on the planet is not entirely science fiction. Already, new technologies are allowing significant fractions of the genetic code to be re-read and checked for spelling differences in large numbers of individuals. It is not far-fetched to imagine that early in the next century this will become possible for the entire genome—although probably not with the ease portrayed in the recent film GATTACA. Even today, the thought experiment of what we can learn from this information places an upper bound on what can be done with more limited data.
The central question posed above has many facets. One concerns the genetic basis of disease. Genetic factors are thought to play some role in susceptibility to almost all diseases. Which spelling differences (muta-
tions) in the genetic code cause or increase susceptibility to which diseases? We can attack this problem using several approaches. Perhaps the most well-known entails tracing inheritance patterns of disease in families in order to implicate particular chromosomal regions in disease transmission. These regions can then be scoured for the causative mutations. Another approach is to start with a mutation and examine its effect on frequency of disease in populations. Alternatively, we can recognize that all people share ancestry sufficiently far back in time, and that many mutations in today’s population are likely to have a common origin. We can therefore search the DNA of population members for ancestral signatures of this origin.
These approaches can be extended from studies of disease to those of normal human variation. They can also be applied to other species, including plants and animals of agricultural importance, model organisms of medical or biological interest, and pathogens.
Another facet concerns the evolutionary history of human populations. It appears likely that all of today’s populations descend from a single, small ancestral population that lived on the plains of East Africa roughly 100,000 years ago. The pattern of spelling differences in our DNA contains signatures of subsequent population divergences, migrations, bottlenecks, replacements, and expansions. If we can understand how to read these signatures, we can answer many interesting questions about our history, as well as about the genetic diversity of and the relationships among today’s populations. In addition to their inherent interest, the answers provide important insights into the use of populations for studies of the genetic basis of disease, including the specific advantages offered by different populations.
The genetic code is a static record of information. It alone does not tell us about the dynamic circuitry of a cell. The response of a cell to changing conditions is governed in large part by which genes are expressed—transcribed into messenger RNA. Understanding the regulatory networks of gene expression is thus a key part of studying any important biological process.
A final facet concerns our responsibilities in dealing with the consequences of genetic research. The key responsibility is education—we must make our research and its implications clearly and broadly understood. Armed with appropriate information, society will be better prepared to face the complex questions posed by progress in human genetics, reaping the benefits while avoiding the pitfalls.
With this survey of the landscape, I now examine the different facets in turn.
GENETIC BASIS OF DISEASE IN FAMILIES
Many diseases run in families. The list includes both diseases with clear genetic causes and simple patterns of inheritance, such as cystic fibrosis and Huntington’s disease, and diseases with more subtle genetic predispositions, such as heart disease and schizophrenia. To date, most of the progress in our understanding of the genetic basis of disease has come from tracing inheritance patterns in families in order to implicate particular chromosomal regions, and ultimately to identify the culprit genes.
Linkage Studies: The Principle
Family studies rely on the principle of genetic linkage, first elucidated by Sturtevant and Morgan in their work on the fruit fly Drosophila in the early part of this century. Suppose a disease gene is segregating in a family, with all affected members inheriting a mutated copy from a single ancestor. As the chromosome on which the mutation resides is transmitted from one generation to the next, regions close to the mutation tend to be inherited with it, while those further away frequently recombine, as illustrated in Figure 1. A correlation is thereby established between genetic markers near the mutation and the mutation itself. One can therefore determine the general location of the disease-causing gene by looking for markers whose transmission correlates with inheritance of disease. Once a gene is thus localized, its position can be further refined by a range of techniques, leading eventually to identification of the gene itself and the disease-causing mutations.
Linkage Studies: Genetic Markers
In Drosophila, simple Mendelian characteristics such as eye color serve as genetic markers. A catalog of many such characteristics allows straightforward genetic localization of new phenotypes using crosses. This approach does not work in humans because we cannot set up crosses, and there are few common polymorphic characteristics (blood type and immunocompatibility loci providing some important exceptions). For many years, this lack of genetic markers hampered the usefulness of linkage for mapping human disease genes—the theoretical principle was waiting for the molecular tools to catch up. The situation changed dramatically with the proposal in 1980 by David Botstein and colleagues that spelling changes in DNA, which are highly abundant, can be used as markers (Botstein et al., 1980). Genetic maps covering all the chromosomes were
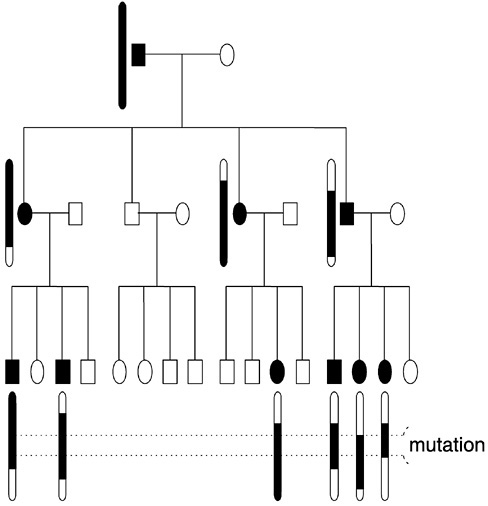
FIGURE 1 Transmission of a dominant disease gene through a family. For clarity, only the mutation-bearing chromosomes in affected individuals are shown. Shaded segments represent pieces of the chromosome on which the mutation entered the family. Dotted lines delimit the region to which the mutation can be confined based on the information in this family.
rapidly developed, and we have seen the ensuing progress in mapping and cloning Mendelian disease genes. Genes for over 700 diseases have been localized to chromosomal regions, and over 100 of these have been identified, including genes involved in cystic fibrosis, Huntington’s disease, muscular dystrophy, and hemochromatosis.
Common Diseases: Genetic Complexity
Mendelian diseases are rare. Even the most common ones such as cystic fibrosis affect no more than one in a thousand people. Many others occur only in a handful of families. In total, Mendelian diseases are thought to affect roughly 1 percent of the population. By contrast, diabetes alone affects about 2 percent of people worldwide. Diabetes has a strong genetic component: A child of a diabetic faces a fivefold increased risk of developing diabetes. Genetic factors also play an important role in other common diseases, such as heart disease, cancer, multiple sclerosis, asthma, manic depression, and schizophrenia. Clearly, genetics can have its biggest impact on medicine by improving our understanding of common disease causation. Yet progress in this area has been modest compared with that in the study of Mendelian diseases. Why?
In the case of common diseases, the linkage approach runs into the problem of genetic complexity. In simple Mendelian diseases, mutations in a single gene always lead to disease and are responsible for all cases of the disease. In the case of genetically complex phenotypes, which encompass virtually all common diseases, this perfect correlation between genotype and phenotype breaks down in one or more of several ways. First, mutations in a gene may lead to disease only some of the time, depending on environmental, other genetic, or random factors. Second, some disease cases may reflect nongenetic causes. Third, mutations in any one of several genes may cause the same disease, or mutations in multiple genes may be required for disease causation. As a result of these and other problems, linkage studies of common diseases are much more difficult than those of simple Mendelian disorders.
One approach to overcoming the problems posed by complexity is to look for families in which a common disease approximately shows Mendelian single-gene inheritance. A prominent example of this approach is the successful identification of breast and ovarian cancer genes BRCA1 and BRCA2. However, this approach may not tell us about the genetic basis of a disease in the general population. Also, families with simple inheritance have not been found for many important diseases.
Common Diseases: Interpretation of Results
Despite the difficulties outlined above, recent years have seen frequent announcements that a gene “for” schizophrenia or alcoholism or reading disability has been found. Many of these findings have not stood the tests of time and replication, whereas others remain a subject of contention. Why has it been so difficult to establish whether a correlation
between a chromosomal region and a common disease is real or a statistical artifact? In simple Mendelian disorders, we know that a single gene accounts for all cases of disease. In the case of common diseases, the number of susceptibility genes and the magnitude of their effects are unknown prior to analysis. A typical study employs a very large number of statistical tests in screening the entire genome for a possible effect. Smaller effects are obscured by random fluctuations in inheritance. As a result, the “signal” is easily confused with “noise.”
To ease the problem of interpretation, Eric Lander and I developed guidelines for reporting results from family studies (Lander and Kruglyak, 1995). The guidelines attempt to strike a balance between avoiding too many false results and not missing true effects. A critical application of the guidelines allows us to provide a realistic picture (albeit with blurry edges) of what we really know about the genetic basis of common diseases. New reports on CNN that a gene for disease x has been identified can be treated with appropriate caution.
Common Diseases: Unknown Mode of Inheritance and Small Effects
A simple Mendelian disorder shows recessive, dominant, or sex-linked inheritance. By contrast, the mode of inheritance of common diseases is typically unknown. An incorrect guess about the mode of inheritance can cause a gene to be missed. Genes involved in common diseases also have relatively small effects on overall disease susceptibility. Detection of such effects requires studies with much larger sample size, placing greater importance on efficient analysis of the data. Over the past several years, a large part of my research effort has focused on providing human geneticists with more powerful tools for detecting complex genetic effects (Kruglyak and Lander, 1995, 1998; Kruglyak et al., 1995, 1996). I have developed statistical techniques that can detect susceptibility genes without having to make guesses about the unknown mode of inheritance. I have also developed computer algorithms and software that allow much more efficient and rapid analysis of family studies. Unlike earlier methods that focused on single genetic markers, the new methods are explicitly designed to take full advantage of the greater power of today’s dense genetic maps. My computer program GENEHUNTER has been used to find chromosomal regions that may be involved in a number of diseases, including diabetes, schizophrenia, inflammatory bowel disease, multiple sclerosis, and prostate cancer.
Interlude: Genetics of Mosquito Resistance to the Malaria Parasite
Linkage approaches can also be used in nonhuman organisms to study a number of important problems. Malaria affects hundreds of millions of people worldwide, primarily in developing countries. The parasites that cause malaria are transmitted to humans by mosquitoes, within which they must spend a stage of their life cycle. Thus, one approach to combating malaria is to interrupt the chain of transmission by making mosquitoes resistant to the parasites. Some mosquitoes are naturally resistant, whereas others are susceptible, with genetic factors playing a clear role in this variation.
Together with Ken Vernick, Yeya Toure, and Fotis Kafatos, we have launched a project to study the genetic basis of parasite resistance in the mosquito population of Mali. In the study, wild female mosquitoes are caught in a local village and allowed to lay eggs in captivity. Lab-reared progeny are then infected with natural populations of the malaria parasite and examined for resistance or susceptibility. Once this phase is completed, we will search for linkage between genetic markers and parasite resistance in mosquito families. The study is similar to family human studies in that mosquitoes are drawn from an outbred population, rather than from inbred laboratory strains. This design allows detection of genetic factors that are important in the wild. An important advantage of the mosquito system, which should help overcome the genetic complex-ity of parasite resistance, is the ease of generating very large families—a single female mosquito can produce over 100 progeny. The results of the study will contribute to a better understanding of the mosquito-parasite interaction and may ultimately permit the development of a genetic strategy to interfere with malaria transmission.
Summary
Family studies have produced many important advances in our understanding of the genetic basis of diseases and other phenotypes, particularly those with simple Mendelian inheritance. Although some tantalizing hints regarding the genes involved in more common disorders have emerged from family studies, successes have been harder to come by because of the problems posed by genetic complexity. It is now becoming increasingly clear that the genetic basis of common diseases is likely to consist of a large number of subtle effects, which interact with each other and the environment. Detection of such effects by linkage requires a prohibitively large number of families. Population studies may offer a way to overcome this hurdle.
GENETIC BASIS OF DISEASE IN POPULATIONS: COMMON VARIANTS
Example: Alzheimer’s Disease
Alzheimer’s disease is a devastating neurological disorder that strikes about 1 percent of people aged 65-70 and up to 10 percent of people over 80 years of age. Its prevalence is on the rise as the population ages. Progress in understanding and treatment of Alzheimer’s disease is thus a key step in extending healthy life span. Family studies have identified several genes that cause early-onset Alzheimer’s disease in some families, but these genes contribute to only a small fraction of all cases. What are the genetic components of the much more common late-onset disease?
Several lines of evidence led to the Apolipoprotein E (APOE) gene. This gene has three common variants, or alleles: E2, E3, E4, each one differing by a single nucleotide. Studies of the APOE gene in Alzheimer’s patients and healthy controls showed that although the control frequency of the E4 allele is about 15 percent, it goes up to 40 percent in patients. Each copy of the E4 allele raises a person ’s risk of Alzheimer’s disease roughly fourfold. Variation at the APOE locus appears to account for about 50 percent of the total contribution of genes to Alzheimer’s disease. Interestingly, variation in the APOE gene also contributes to cholesterol levels and heart disease susceptibility, and may play a role in longevity.
Association Studies: Basic Concept and Advantages
The APOE-Alzheimer’s story is an illustration of the simplest type of population studies, known as association studies. These studies directly examine the effect of a particular variant on a disease (or other phenotype). In the future, such studies will be conducted by resequencing entire genomes and looking for consistent differences between patients and controls. Until this becomes feasible, the strategy is to find common variants in genes and examine their phenotypic effects in populations. The approach therefore relies on the existence of such common variants, as well as on our ability to detect them.
Association studies have important advantages over family linkage studies. Instead of searching for an indirect correlation between linked markers and a phenotype, one can look directly at the effect of a particular gene variant on a phenotype. As a result, the power to detect subtle genetic effects is much higher, extending to effects that are undetectable by family studies. It is also much easier to find and recruit individual patients and controls than it is to collect extended families segregating a disease. The results of association studies—in particular, measures of risk
conferred by a gene variant—are more directly applicable to the general population.
Association Studies: Interacting Genes
A polymorphism at one locus may have no effect on susceptibility by itself, but it may have a significant effect when coupled with a particular polymorphism at another locus. An illustration is provided by the system of enzymes that metabolize drugs, carcinogens, and other foreign chemicals. This metabolism is accomplished in two phases. First, a phase I enzyme attaches an oxygen to the chemical to form a reactive intermediate. Next, a phase II enzyme uses this oxygen to add a soluble group, which allows the removal of the chemical from the body. The intermediate compound is often toxic or carcinogenic. A mismatch in the rates of the two enzymes can lead to an accumulation of this compound, with obvious adverse consequences. For example, there is evidence that a particular allele of the phase I enzyme cytochrome P450 1A1, combined with a particular allele of the phase II enzyme glutathione-S-transferase, leads to a ninefold increase in lung cancer risk among Japanese smokers (Hayashi et al., 1993). Studies in mice have also shown that susceptibilities to both lung and colon cancer are influenced by interacting genes (Fijneman et al., 1996).
More complex interactions among multiple loci are also possible, and there is every reason to expect that they are the rule rather than the exception. How can we detect such effects? One approach is to examine directly the phenotypic effects of all possible allelic combinations at sets of loci. Although this may be feasible (if challenging) for pairs of loci, the number of combinations quickly becomes intractable when we consider larger sets.
An alternative is to look for correlations among polymorphisms in the general population. Selection over evolutionary time scales is likely to have favored particular combinations of polymorphisms at loci that jointly affect a phenotype, whereas variation at loci with independent phenotypic effects should be uncorrelated. The task is to uncover meaningful correlations among polymorphisms by analyzing the frequency of their joint occurrence in many individuals. Sets of interacting polymorphisms discovered in this fashion can then be tested for specific phenotypic effects. If successful, this two-stage approach offers greater power to detect subtle genetic effects by drastically limiting the number of combinations tested in the second stage.
Association Studies: Technical Considerations
Study Size
Standard statistical methods can be used to show that a sample of roughly 100 cases and 100 controls is sufficient to detect common polymorphisms that increase the risk of a disease fourfold—roughly the effect of the E4 allele on Alzheimer’s—whereas a sample of about 1,000 is needed for polymorphisms that increase the risk twofold. Lesser risks require increasingly large samples, and practical constraints on sample collection and molecular tests will determine how small an effect can be detected. It is interesting to ask: What gene effects on phenotypes are too subtle to be detected even given the complete DNA sequence of every person on the planet?
Number of Alleles
Effect size depends on the number of functional alleles at a locus. Even if a locus contributes strongly to a phenotype, this contribution may be split among so many different polymorphisms that the effect of any one allele is undetectable. It is thus important to ask under what circumstances we can expect reduced allelic complexity. For instance, are there many fewer alleles in genetically isolated populations? This question is as yet largely unexplored.
Multiple Testing
When many polymorphisms at many loci are tested against many phenotypes, we run the risk of being swamped by false-positive results that will inevitably occur in such a fishing expedition. How do we combat this problem? There are indications that quite strict statistical criteria can be applied without too onerous an increase in sample size, but more precise guidelines need to be formulated.
Population Substructure
False associations will also show up when both a disease and a polymorphism have different frequencies in different ethnic subgroups of a population. Such substructure can be a problem even within broad racial categories such as “Caucasian.” The problem can be overcome by using family members as controls, but remains an issue for data sets in which family members are not available. This is the case with many valuable population-based samples that have been collected over the years, for example the Framingham study.
Summary
Association studies directly address the genetic basis of disease in populations. They are quite powerful and require relatively few assumptions. Ultimately, this approach will dominate in studies of the relationship between genotypes and phenotypes. In the shorter term, the approach faces technical challenges. A systematic search of the genome by direct association involves tests of up to several variants in each of roughly 100,000 genes. It also requires the variants to be known, which may be difficult in the case of noncoding polymorphisms that alter the expression level of a gene by changing a control region or by interfering with the processing of messenger RNA. Until all variants are known and it is feasible to test them, studies based on ancestral signatures of common mutations may offer a shortcut.
GENETIC BASIS OF DISEASE IN POPULATIONS: ANCESTRAL SIGNATURES
Although some diseases such as hemophilia are frequently caused by new mutations, many important disease mutations arose only once or a few times in human history. For example, although many mutations in the cystic fibrosis transmembrane conductance regulator gene can cause cystic fibrosis, a single mutation of unique origin, known as ∆F508, is responsible for 75 percent of the cases. Similarly, the sickle cell mutation in the hemoglobin gene is likely to have arisen between one and four times. We know this because of ancestral signatures in the DNA surrounding a mutation.
Every mutation must arise at a particular time in a particular person —some baby born in Africa thousands of years ago carried the first sickle cell mutation. A mutation arises on a particular chromosome, with a unique signature of spelling changes. As generations pass, the signature is eroded by recombination, but remains largely intact in a region immediately surrounding the mutation, as illustrated in Figure 2.
Such a signature can serve as a proxy for knowing the mutation itself and can be tested for its phenotypic effect. This approach, known as linkage disequilibrium (LD) mapping, has been instrumental in narrowing the location of a number of Mendelian disease genes, including those for cystic fibrosis (Kerem et al., 1989) and diastrophic dysplasia (Hastbacka et al., 1992). The use of LD mapping for common diseases, as well as for initial gene detection, is largely untested. This situation is expected to change shortly as new technologies begin to allow systematic searches of the genome for ancestral signatures.
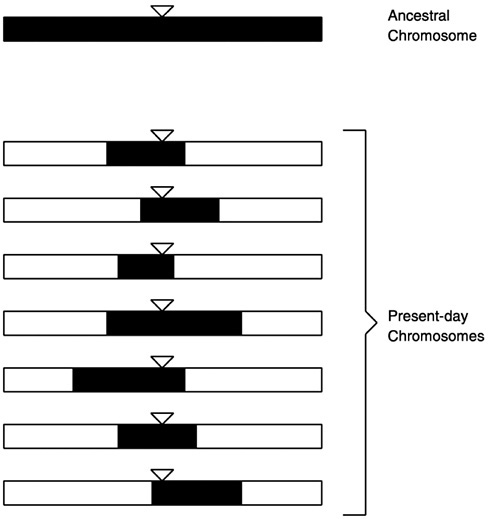
FIGURE 2 Signature of an ancestral mutation in present-day chromosomes. The ancestral mutation is denoted by the triangles. Shaded segments of present-day chromosomes retain the ancestral signature.
Example: Familial Mediterranean Fever
Familial Mediterranean fever (FMF) is, as the name suggests, a disease of inflammation that runs in families of Mediterranean origin. FMF patients suffer from periodic bouts of fever accompanied by other symptoms. Although FMF is rare in most populations, its frequency reaches 1 percent among non-Ashkenazi Jews, Armenians, Arabs, and Turks. This observation raises interesting questions about a possible selective advan-
tage for carriers in the Mediterranean region and what the selective agent might be.
FMF shows simple recessive inheritance and is particularly common in offspring of consanguineous marriages. Working with Dan Kastner and colleagues, we used linkage to localize the FMF gene to a region of roughly four million base pairs on chromosome 16. Although this stretch of DNA comprises only a tenth of 1 percent of the entire genome, it is still too large for a systematic gene search. To narrow the location further, we searched for an ancestral signature in the North African Jewish population. The pattern of spelling changes in the DNA of patient chromosomes indicated that over 90 percent of them descend from a single ancestral chromosome on which the original FMF mutation arose. Lining up the ancestral segments (as illustrated in Figure 2), we were able to confine the gene’s location to several hundred thousand base pairs (Levy et al., 1996), a region small enough to permit the identification of the gene, which was announced recently (International FMF Consortium, 1997). I return to the FMF story below.
Ancestral Signatures: Technical Considerations
Existence of Ancestral Signatures
LD between a functional polymorphism and surrounding markers must first be established in a population. It then needs to be maintained for many generations over appreciable chromosomal distances, without being eroded beyond recognition by recombination, mutation, and other factors. Under what circumstances are these conditions likely to be met, and how universal a phenomenon is LD? Except for young isolated populations, only limited theoretical guidance—and very little data —currently exists to begin answering this question.
Chromosomal Extent of Ancestral Signatures
Some mutations are old, arising when our ancestors wandered the plains of East Africa 100,000 years ago. Recombination will have had 5,000 generations to whittle away their ancestral signatures. Other mutations are more recent, arising in Finland 2,000 years ago, or being introduced into Canada by French settlers in the seventeenth century. These will be surrounded by much larger ancestral signatures. The density of genetic markers required to detect ancestral signatures thus depends on a mutation’s history. Theoretical arguments can be used to estimate that a systematic search of the genome for LD in old mixed populations (such as that of the United States) will require roughly 500,000 markers (Kruglyak,
1999). Better empirical data are needed to accurately address this question.
Background Noise
Shared ancestral segments can occur by chance. The extent of such background sharing is not well characterized. A better description of this “noise” is essential for us to be able to recognize the signal of sharing related to phenotype.
Mutations with Multiple Origins
A particular mutation may arise more than once in the history of a population. It will then occur on chromosomes with several different ancestral signatures. For example, early-onset torsion dystonia is caused by a deletion of three nucleotides that appears to have occurred multiple times in different ethnic groups (Ozelius et al., 1997). In such cases, no correlation can be found between any one ancestral signature and disease. We need to have a better idea of how common this situation is.
Summary
An approach based on ancestral signatures has the advantage of detecting effects that are due to previously unknown variants. This approach would be very attractive if a reasonable number of markers sufficed to adequately cover the genome. However, as seen above, this is unlikely to be the case in general populations—around 500,000 markers may be required. The necessary number of markers can be smaller for young (recently founded) isolated populations, although only under a narrow set of conditions (Kruglyak, 1999). LD mapping also relies on a number of assumptions that are needed to ensure that ancestral signatures are established and maintained. Validating these assumptions requires greater knowledge of population structure and history—and their impact on allelic complexity of different populations and on the extent of ancestral signatures. Obtaining such knowledge is the subject of the next section.
POPULATION HISTORY TRACES OF HUMAN EVOLUTION IN DNA
Example: Familial Mediterranean Fever
I now return to the story of FMF. As noted above, ancestral signatures played a key role in fine localization and eventual identification of
the FMF gene. When the disease-causing mutations in the gene were identified, surprises emerged (International FMF Consortium, 1997). Over 80 percent of patient chromosomes carry one of four mutations, each changing a single amino acid. Patterns of spelling changes in the surrounding DNA strongly suggest that each mutation arose only once. Strikingly, patients of Armenian, Ashkenazi Jewish, and Druze origins share the same mutation and the surrounding ancestral signature, indicating common ancestry of populations that have been physically separated for over 2,000 years and were previously thought to be unrelated. Another mutation is found in Jewish patients of both North African and Iraqi origins, even though the latter group has been isolated from the rest of the Jewish population since the Babylonian captivity of 2,500 years ago. Similar unexpected relationships were found among other Mediterranean populations. We are currently extending these studies to learn more about the history and interrelatedness of the region’s people.
Reconstructing History from Molecular Data
The example of FMF raises a general question: How do we reconstruct the history of human populations? Diversity of individual populations and relationships between populations are of inherent interest. As noted above, they also provide important information about the suitability of a population for finding disease genes.
We could begin the reconstruction by examining written records, oral histories, and mythologies. However, such records for most populations are incomplete at best, and even in the case of unusually well-characterized populations, such as Icelanders or the Amish, much of the information about population diversity still needs to be either discovered or confirmed empirically. If a population uses names that are passed from generation to generation in a systematic fashion, we could examine the number and frequency of different names. Such “last name analysis ” has actually been used with success to assess the genetic diversity of some genetically isolated populations, including the Hutterites and French Canadians, but it cannot be used to establish relationships among populations or to shed light on the chromosomal extent of ancestral signatures.
The best repository of historical information is the DNA of population members. Patterns of spelling changes in the genetic code contain a record of population divergences, migrations, bottlenecks, replacements, and expansions. Indeed, molecular approaches have already contributed a great deal to our understanding of human evolution. A prominent example is provided by the studies of mitochondrial DNA that led to the “mitochondrial Eve” hypothesis of a recent African origin of modern humans. These approaches need to be updated to take full advantage of modern molecular tools, provide a more detailed look at the entire ge-
nome, and resolve finer points of population diversity and relationships. In the future, complete DNA sequences will be used for this purpose. In the meantime, we must rely on sampling polymorphisms from genomes of population members. Given such data, we can observe the number of different ancestral signatures present in our sample, their frequency distribution, and the range of chromosomal distances over which they extend. Such observations contain information about a number of historical parameters of the population, including its age, number of founders, rate of expansion, and bottlenecks (population crashes). For example, the presence of extended ancestral signatures indicates that a population is of recent origin, whereas the existence of only a few ancestral signatures indicates that a population has limited genetic diversity, likely due to a small number of founders or a bottleneck since the founding of the population.
Reconstructing History: Technical Considerations
Which Markers?
Several options are available. Microsatellite repeats, currently the most commonly used genetic markers, are highly polymorphic, but mutate at high frequency. Single nucleotide changes are highly abundant in the genome, more stable over time, but less polymorphic. Large insertions and deletions, most likely to represent unique mutational events in the history of the population, are relatively rare. These different types of markers may each have a role to play in answering different questions, depending on the relative importance of the polymorphism level (which allows us to distinguish different ancestral signatures), mutational stability (which allows us to look far back in time), and abundance (which may be needed to adequately sample the genome).
Marker Location
Most simply, markers can be chosen in random genomic regions, where they can be used to examine the extent of LD and the haplotype diversity in a population. Alternatively, they can be chosen around already known functional polymorphisms—such as those in the APOE gene associated with Alzheimer’s disease—where their usefulness in detecting association can be tested.
Population Sample
Given an appropriate set of markers, we need to know how many members of the population must be included in the study sample, as well as how they should be chosen. One to two hundred independent chro-
mosomes (or fifty to a hundred people) should allow a reasonably accurate estimation of population parameters, although the exact numbers will need to be computed for each particular task. The numbers will also vary from population to population, with more diverse, older populations likely to require larger samples for accurate characterization.
Summary
Reconstructing population history is one of the traditional goals of population genetics, and tools and insights from that field will undoubtedly be useful. The traditional approaches will need to be refocused from broad evolutionary questions about human origins to finer-scale population characteristics and relationships, particularly those that are relevant to searches for disease genes. They will also need to be updated to handle the amount and type of information that can be readily generated by modern molecular techniques. Finally, we can pose a provocative question: What aspects of human history cannot be reconstructed even from the complete DNA sequence of every person on the planet and are thus fundamentally unknowable (at least without a time machine)?
BEYOND DNA SEQUENCE: REGULATORY NETWORKS
DNA sequence is a static record of genetic information. The dynamic response of a cell to changing conditions is governed in large part by which genes are expressed—transcribed into messenger RNA. Understanding the regulation of gene expression is thus a key part of studying any important biological process.
Several emerging technologies now allow simultaneous measurements of transcription levels of many genes in a cell under a variety of conditions. It is then relatively straightforward to identify genes whose levels of expression under two conditions differ strongly. A more general—and difficult—task is to reconstruct the network of interactions among genes from these observations. I conjecture that this network will reveal distinct modules of genes whose expression levels correlate strongly within a module but more weakly between modules. Evidence of such modules has been found in a study of p53-mediated apoptosis (Polyak et al., 1997), as well as in a study of metabolic shift from fermentation to respiration in yeast (DeRisi et al., 1997). A particularly interesting question then arises: Do cells respond to changes in conditions simply by turning modules on and off or by reconfiguring the entire system in novel ways?
The budding yeast Saccharomyces cerevisiae provides a well-suited system for investigating this question, because the expression of essentially
all genes can be monitored simultaneously under a broad range of precisely controlled growth conditions. Preliminary calculations suggest that a reasonable number of whole-genome expression measurements, each carried out under different growth conditions, suffices to untangle real correlations between pairs of genes from random noise. These pairwise correlations can then be examined further to uncover networks of interactions among larger collections of genes. I am currently planning computer simulations to test these ideas. I have also established collaborations with several yeast biologists to develop experiments aimed at addressing the question experimentally. Insights drawn from yeast can then be applied to more complex systems such as human tumorigenesis.
Dynamic responses of a cell are not confined to changes in the levels of gene expression. Many important processes are governed by modifications and interactions of proteins. Technologies to monitor levels of proteins, to distinguish different modifications of a protein, and to detect interactions between proteins are improving rapidly. Eventually, these technologies will allow a global examination of all proteins in the cell. Ideas developed to understand networks of interacting genes can then be applied to this next level of complexity.
THE ROLE OF THEORY
Theoretical thinking has a long intellectual tradition in biology. Reconciling the evolutionary ideas of Darwin with the Mendelian concept of particulate inheritance required the landmark quantitative efforts of Fischer, Wright, and Haldane. Theoreticians such as Delbruck, Crick, and Gamow made essential contributions in the early days of molecular biology (Judson, 1996). Today, as we face the challenge of understanding complex biological systems, theory has an important role to play.
Numerous complex systems are characterized by having many functional units with dense networks of interactions and by the need to infer underlying structure from incomplete, noisy observations. As a result, they pose conceptually similar problems. For example, the approach to understanding networks of gene expression described above draws on lessons and insights from work on information processing by large assemblies of neurons. The same ideas can be applied to understanding correlated genetic variation in populations.
A fruitful theoretical approach to complex systems needs to be firmly grounded in the data in order for the results to be applicable to the real world. At the same time, it should do more than simply react to the data. Indeed, the most valuable contribution of theory is to guide the field by making predictions and suggesting novel experiments.
The cultures of research, and not just techniques, differ between fields.
Delbruck, Crick, and Gamow—physicists entering molecular biology—brought with them a spirit of free information exchange, infectious enthusiasm, and easy speculation, evident in the informal scientific networks of the Phage Group and the RNA Tie Club (Judson, 1996). I hope to foster a similar intellectual atmosphere in modern human genetics, both within my research group and in the broader research community.
EDUCATION: A KEY RESPONSIBILITY
Public Perception of Genetic Research
The current level of public understanding of genetics is distressingly low. This is both contributed to and illustrated by coverage in the popular media—with headlines such as “Your whole life is in your genes at birth” from the tabloid World Weekly News, “Gene mapping: A journey for tremendous good—or unspeakable horrors” from The Village Voice, and “Couples can order embryos made to their specifications” from the New York Times. The recent science fiction film GATTACA is set in a “not-too-distant future” world in which every child’s likely fate, including the time and cause of death, is predicted at birth from DNA analysis. Such portrayals tend to overstate greatly the degree to which genes determine physiology and behavior. They also frequently focus on extreme examples of genetic technologies, such as human cloning. As a result, they run the twin risks of, on the one hand, a backlash against all applications of genetics, no matter how beneficial, and on the other hand, a lack of preparation for the hard choices that lie ahead.
The Role of Education
I believe that our most important responsibility in dealing with the consequences of our research is education. Scientists cannot make choices for society—although we can, and should, avoid and discourage lines of research that we find unethical. Instead, we must make our research and its implications clearly and broadly understood. The public needs to know that we will never be able to make accurate predictions based on DNA sequence about many important aspects of human life, such as time and cause of death, level of accomplishment, and happiness. Similarly, although the medical benefits of human genetics are considerable, they should not be oversold, especially in the short term. Progress in understanding a disease—and especially in developing a treatment—can take many years, even after we know its genetic basis. Excessive emphasis on the medical benefits also short-changes the purely intellectual rewards of greater knowledge of human biology and evolutionary history.
Armed with appropriate information, society will be better prepared to face the complex questions posed by progress in human genetics, reaping the benefits while avoiding the pitfalls.
CONCLUSION
As human genetics enters the twenty-first century, many powerful molecular tools are either in place or about to arrive. They include a large capacity for DNA sequencing, an ability to examine many polymorphisms in many people, and techniques for monitoring expression levels of many genes under many conditions. These tools are already beginning to generate an avalanche of data. Early in this century, we can envision obtaining complete DNA sequences of many individuals, expression levels of all genes in a variety of cells, and a complete characterization of protein levels and interactions. The key task facing human genetics is interpreting this data.
In this chapter, I have tried to sketch out some of the intellectual tools that are needed for this task. The description is certainly not exhaustive, and some needs cannot be anticipated today. It is instructive to recall that the polymerase chain reaction is just over a decade old, but has had a profound impact on virtually all aspects of molecular genetics—it is difficult to imagine how most current approaches would be feasible without it. New technologies of similar power are certain to come along. Flexibility and openness to new ideas are thus of the essence. A unifying thread is provided by the realization that quantitative thinking will always be a necessary complement for experiments carried out on a global genomic scale.
Success in the task of interpretation will clarify the genetic basis of human diseases and other phenotypes. It will also provide insights into interesting aspects of population history. The general approach extends beyond human genetics to studies of pathogens and organisms of agricultural importance, and even more broadly to a better understanding of genetics and biology of all organisms. Thus, the ultimate benefits to society take many forms: better medicine, improved quality of life, increased knowledge, and a greater understanding of who we are and where we fit in among other forms of life.
REFERENCES
Botstein, D., D. L. White, M. Skolnick, and R. W. Davis. 1980. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. American Journal of Human Genetics 32:314–331.
DeRisi, J. L., V. R. Iyer, and P. O. Brown. 1997. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science 278:680–686.
Fijneman, R. J., S. S. de Vries, R. C. Jansen, and P. Demant. 1996. Complex interactions of new quantitative trait loci, Sluc1, Sluc2, Sluc3, and Sluc4, that influence the susceptibility to lung cancer in the mouse [see comments]. Nature Genetics 14(4):465–467.
Hastbacka, J., A. de la Chapelle, I. Kaitila, P. Sistonen, A. Weaver, and E. Lander. 1992. Linkage disequilibrium mapping in isolated founder populations: Diastrophic dysplasia in Finland. Nature Genetics 2:204–211.
Hayashi, S. I., J. Watanabe, and K. Kawajiri. 1993. High susceptibility to lung cancer analyzed in terms of combined genotypes of P450IA1 and mu-class glutathione S-transferase genes. Japanese Journal of Cancer Research 83:866–870.
International FMF Consortium. 1997. Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell 90:797–807.
Judson, H. F. 1996. The Eighth Day of Creation, expanded ed. Cold Spring Harbor, N.Y.: CSHL Press.
Kerem, B., J. M. Rommens, J. A. Buchanan, D. Markiewicz, T. K. Cox, A. Chakravarti, M. Buchwald, and L. C. Tsui. 1989. Identification of the cystic fibrosis gene: Genetic analysis. Science 245(4922):1073–1080.
Kruglyak, L. 1999. Prospects for whole-genome linkage disequilibrium mapping of common disease genes. Nature Genetics 22:139–144.
Kruglyak, L. and E. S. Lander. 1995. Complete multipoint sib pair analysis of qualitative and quantitative traits. American Journal of Human Genetics 57:439–454.
Kruglyak, L. and E. S. Lander. 1998. Faster multipoint linkage analysis using Fourier transforms. Journal of Computational Biology 5:1–7.
Kruglyak, L., M. J. Daly, and E. S. Lander. 1995. Rapid multipoint linkage analysis of recessive traits in nuclear families, including homozygosity mapping. American Journal of Human Genetics 56:519–527.
Kruglyak, L., M. J. Daly, M. P. Reeve-Daly, and E. S. Lander. 1996. Parametric and nonparametric linkage analysis: A unified multipoint approach. American Journal of Human Genetics 58:1347–1363.
Lander, E., and L. Kruglyak. 1995. Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nature Genetics 11(3):241-247.
Levy, E. N., Y. Shen, A. Kupelian, L. Kruglyak, I. Aksentijevich, E. Pras, Je Balow, Jr., B. Linzer, X. Chen, D. A. Shelton, D. Gumucio, M. Pras, M. Shohat, J. I. Rotter, N. Fischel-Ghodsian, R. I. Richards, and D. L. Kastner. 1996. Linkage disequilibrium mapping places the gene causing familial Mediterranean fever close to D16S246. American Journal of Human Genetics 58(3):523–534.
Ozelius, L. J., J. W. Hewett, C. E. Page, S. B. Bressman, P. L. Kramer, C. Shalish, D. de Leon, M. F. Brin, D. Raymond, D. P. Corey, S. Fahn, N. J. Risch, A. J. Buckler, J. F. Gusella, and X. O. Breakefield. 1997. The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nature Genetics 17:40–48.
Polyak, K., Y. Xia, J. L. Zweier, K. W. Kinzler, and B. Vogelstein. 1997. A model for p53-induced apoptosis. Nature 389:300–305.