Reference Manual on Scientific Evidence: Fourth Edition (2025)
Chapter: Reference Guide on Human DNA Identification Evidence
Reference Guide on Human DNA Identification Evidence
David H. Kaye, M.A., J.D., is Regents Professor Emeritus, Arizona State University Sandra Day O’Connor College of Law and School of Life Sciences, and Distinguished Professor of Law and Academy Professor Emeritus, Pennsylvania State University School of Law.
Author’s Note: Research for this reference guide was completed in 2023. I am grateful not only to the National Academies of Sciences, Engineering, and Medicine and Federal Judicial Center reviewers and staff, but also to Bruce Budowle, John Butler, Michael Coble, David Housman, Jarrah Kennedy, Swathi Kumar, and Bruce Weir for their comments on portions of a draft of this reference guide.
CONTENTS
A Brief History of DNA Evidence
Variation in Human DNA and Its Detection
What Are DNA, Chromosomes, Genes, Proteins, and RNAs?
What are genes, proteins, and RNAs?
How do cells manufacture proteins and RNAs?
Sexual Reproduction and the Genome
What Are DNA Polymorphisms and How Are They Detected?
STRs and Capillary Electrophoresis
Capillary electrophoresis and fluorescence
Miniaturization for rapid capillary electrophoresis
Sequence-Specific Probes and Microarrays
What Establishes That a Genetic System Is Valid for Identification?
Sample Collection and Laboratory Performance
Sample Collection and Contamination
Did the Sample Contain Enough DNA?
Was the Sample of Sufficient Quality?
What Quality Control and Assurance Measures Are in Place?
How Are Samples Created and Handled?
Inference, Statistics, and Population Genetics in Human Nuclear DNA Testing
What Constitutes a Match? An Exclusion?
What Hypotheses Can Be Formulated About the Source?
Can the Match Be Attributed to Laboratory Error?
Could a Close Relative Be the Source?
Could an Unrelated Person Be the Source?
Frequencies, Probabilities, and Prejudice
Are Frequencies or Probabilities Prejudicial Because They Are So Small?
Are Frequencies or Probabilities Prejudicial Because They Might Be Transposed?
“Rarity,” Source, and Uniqueness Testimony
Special Issues in Human DNA Testing
Analytical Methods and Categorical Matches
Population Genetics and Statistics
Haplotype frequency or probability estimates
Mitochondria and Their Genomes
Analytical Methods and Categorical Matches
Have Strategies for Simplifying Mixtures Interpretation Been Pursued?
What Is Mixture Deconvolution?
What Statistical Assessment of the Comparison to a Defendant’s Profile Has Been Conducted?
Probability of inclusion or exclusion
Combined probability of inclusion (CPI) and exclusion (CPE)
Random-match probability (RMP)
Definition of the likelihood ratio
Probabilistic genotyping software (PGS)
DNA and RNA Typing of Bodily Fluids or Tissues
Offender and Suspect Database Searches
Law-enforcement databases and databanks
Probative value of database hits
The statistical analyses of adjustment
Judicial opinions on adjustment
Kinship trawling (“familial searching”)
All-pairs matching within a database to verify estimated random-match probabilities
Investigative Genetic Genealogy (IGG)
How does direct-to-consumer genetic genealogy work?
From the forensic sample to the SNP genotype
From the SNP genotype to possible relatives
Does IGG violate the Fourth Amendment?
Are parts of IGG a search or seizure?
Biogeographic ancestry testing (BGAT)
FIGURES
2. Three alleles of the D16S539 STR
3. Sketch of an electropherogram for two D16S539 alleles
4. Alleles of 15 STR loci and the amelogenin sex-typing test from the AmpFlSTR Identifiler kit
5. Electropherogram for nine STR loci of the victim’s DNA in People v. Pizzaro
TABLE
1. Hypotheses That Might Explain a Match Between Defendant’s DNA and DNA at a Crime Scene
Introduction
Deoxyribonucleic acid, or DNA, is a molecule that encodes the genetic information in all living organisms. Its chemical structure was elucidated in 1953. More than 30 years later, samples of human DNA began to be used in the criminal justice system, primarily in cases of rape or murder. The evidence has been the subject of extensive scrutiny by lawyers, judges, and the scientific community. DNA evidence is admissible in all jurisdictions, but there are many types of forensic DNA analysis, and still more are being developed. Questions of admissibility and weight of DNA evidence arise as advancing methods of biochemical and statistical analysis, along with novel applications of established methods, are introduced. Moreover, questions about the appropriate balance between law-enforcement goals and individual privacy and security also emerge when law-enforcement officials collect and analyze human DNA samples. This reference guide addresses technical issues that are important when considering the admissibility of and weight to be accorded analyses of human DNA and the related molecule RNA (ribonucleic acid) in trials. In addition, this guide describes ways in which human DNA assists in criminal investigations, and it identifies a number of the legal and policy questions raised by investigative genetics.
A Brief History of DNA Evidence
“DNA evidence” refers to the results of chemical or physical tests that directly reveal differences in DNA molecules found in organisms as diverse as bacteria, plants, and animals.1 The technology for establishing the identity of individuals from the DNA in blood, semen, and other bodily fluids became available to law-enforcement agencies in the mid to late 1980s.2 The judicial reception of DNA evidence can be divided into at least six phases.3 The first phase was one of rapid acceptance. Initial praise for RFLP (restriction fragment length polymorphism)
1. Differences in DNA also can be revealed by differences in the proteins that are made according to the “instructions” in a DNA molecule. Blood-group factors, serum enzymes and proteins, and tissue types all reveal information about the DNA that codes for these chemical structures. Such immunogenetic testing, including the application of antibodies to detect cell-surface antigens, predates the more direct DNA testing that is the subject of this guide. On the nature and admissibility of such testing, see, for example, David H. Kaye, The Double Helix and the Law of Evidence 5–19 (2010); 1 McCormick on Evidence § 205(B) (Robert Mosteller ed., 8th ed. 2020). See generally Liesa L. Richter & Daniel J. Capra, Reference Guide on the Admissibility of Scientific Evidence, in this manual.
2. The first reported appellate opinion is Andrews v. State, 533 So. 2d 841 (Fla. Dist. Ct. App. 1988).
3. The description that follows is adapted and updated from 1 McCormick on Evidence, supra note 1, § 205(B). Beyond these six phases for the principal methods of human DNA profiling encountered in courts, the supplementation of STR profiling with other genetic markers for identification related to single-nucleotide differences is beginning.
testing in homicide, rape, paternity, and other cases was effusive.4 Expert testimony rarely was countered, and courts readily admitted DNA evidence.
In a second wave of cases, however, defendants pointed to problems at two levels—controlling the experimental conditions of the analysis and interpreting the results. Concerted attacks by defense experts with impressive credentials led to the rejection of specific proffers on the grounds that the testing was not sufficiently rigorous.5
A different attack on DNA profiling that began in cases during this period led to a third wave of cases in which many courts held that estimates of the probability of a coincidentally matching DNA profile were inadmissible. These estimates relied on a simple population-genetics model for the frequencies of DNA profiles, and some prominent scientists claimed that the applicability of the mathematical model had not been adequately verified. A heated debate on this point spilled over from courthouses to scientific journals and convinced the supreme courts of several states that general acceptance was lacking. A 1992 report of the National Academy of Sciences (NAS) proposed a more “conservative” computational method as a compromise,6 and this seemed to undermine the claim of scientific acceptance of the procedure that was in general use.
An outpouring of critiques of the report led to the formation of a second NAS committee. This panel concluded in 1996 that the usual method of estimating frequencies in broad population groups generally was sound, and it proposed improvements and additional procedures for estimating frequencies in subgroups.7 In the corresponding fourth phase of judicial scrutiny of DNA evidence, the courts almost invariably returned to the earlier view that the statistics associated with DNA profiling are generally accepted and scientifically valid.
4. E.g., People v. Wesley, 140 Misc. 2d 306, 533 N.Y.S.2d 643, 644 (Alb. Cnty. Ct. 1988) (“the single greatest advance in the ‘search for truth’ . . . since the advent of cross-examination”). DNA testing also quickly became powerful evidence of kinship in child support, immigration, and other cases involving genetic parentage or other relationships within a family.
5. Moreover, a minority of courts, perhaps concerned that DNA evidence might be conclusive in the minds of jurors, added a “third prong” to the general-acceptance standard of Frye v. United States, 293 F. 1013 (D.C. Cir. 1923). This augmented Frye test requires not only proof of the general acceptance of the ability of science to produce the type of results offered in court, but also of the proper application of an approved method on the particular occasion. For commentary, see David L. Faigman et al., Modern Scientific Evidence § 30:9 n.5 (2022–2023 ed.) (citing articles); David H. Kaye et al., The New Wigmore, A Treatise on Evidence: Expert Evidence § 7.3.3(a)(2) (3d ed. 2021) (criticizing the approach); Joseph G. Petrosinelli, Comment, The Admissibility of DNA Typing: A New Methodology, 79 Geo. L. J. 313, 327–31 (1990) (similar criticism); cf. Ming W. Chin et al., Forensic DNA Evidence: Science and the Law § 11:6 (2022) (describing California’s “limited third-prong hearing”).
6. National Research Council, DNA Technology in Forensic Science (1992) [hereinafter NRC I].
7. National Research Council, The Evaluation of Forensic DNA Evidence (1996) [hereinafter NRC II].
In the fifth phase of the judicial evaluation of DNA evidence, results obtained with PCR-based methods8 entered the courtroom. The opinions were practically unanimous in holding that the PCR-based procedures rested on a solid scientific foundation and were generally accepted in the scientific community. Before long, forensic scientists settled on the use primarily of one type of DNA variation—known as short tandem repeats, or STRs—to include or exclude individuals as the source of crime-scene DNA. Investigative databases of STR profiles from men, women, and children convicted (and sometimes only arrested for) specified crimes grew by leaps and bounds.9 Matching these recorded profiles to STR profiles of crime-scene samples generated seemingly magical investigative leads.
The sixth phase of judicial scrutiny is still in progress. With the extension of STR profiling to minute quantities of DNA (low template, or LT-DNA), often from more than one individual, laboratories have turned to computerized methods to infer which individual profiles may be giving rise to the patterns of STRs and which features in the data are the result of instrumental error or limitations. Both the modifications for generating data from LT-DNA and the “probabilistic genotyping software” have been the subject of admissibility hearings.
Less frequently, other genetic systems to establish the identity of the source of trace DNA evidence have been applied to generate investigative leads. The highly publicized technique of trawling commercial or other privately maintained databases established for genealogical research, to discover individuals who might be related to the source, is discussed in the section titled “Investigative Genetic Genealogy” below. Inferring visible traits and biogeographic ancestry from DNA found at crime scenes is described in the section titled “Forensic DNA Phenotyping” below. These investigative procedures rely on newer methods for analyzing DNA that are just beginning to be evaluated in court.
Throughout these phases, DNA tests also exonerated an increasing number of people who had been convicted of capital and other crimes, posing serious questions about the adequacy of the legal procedures for obtaining postconviction DNA testing and for overturning factually erroneous convictions.10 The value of
8. PCR (polymerase chain reaction) is described in the section titled “Chromosomes” below.
9. See Maryland v. King, 569 U.S. 435 (2013) (upholding compulsory DNA sampling as part of the booking process for custodial arrests); section titled “Law-enforcement databases and databanks” below. Initially, the law-enforcement databases housed records of restriction-fragment-length polymorphisms arising from variable numbers of tandem repeats (RFLP-VNTR profiles). FBI, Frequently Asked Questions on CODIS and NDIS, https://www.fbi.gov/how-we-can-help-you/dna-fingerprint-act-of-2005-expungement-policy/codis-and-ndis-fact-sheet, last visited Sept. 8, 2024 (“The National DNA Index no longer searches DNA data developed using restriction fragment length polymorphism (RFLP) technology.”).
10. See, e.g., Osborne v. Dist. Atty’s Office for Third Jud. Dist., 557 U.S. 52 (2009) (narrowly rejecting a convicted offender’s claim of a due process right to DNA testing at his expense, enforceable under 42 U.S.C. § 1983, to establish that he is probably innocent of the crime for which he was convicted after a fair trial, when (1) the convicted offender did not seek extensive DNA testing before trial even though it was available, (2) he had other opportunities to prove his innocence after
DNA evidence in solving older crimes also prompted extensions of some statutes of limitations.11
In sum, in little more than a decade, forensic DNA typing made the transition from a novel set of methods for identification to a relatively mature and well-studied forensic technology. However, one should not lump all forms of DNA identification together. New techniques and applications continue to emerge. Before admitting DNA evidence, courts normally inquire into the biological principles and knowledge that would justify inferences from new or different technologies or applications. As a result, this guide describes not only the predominant STR technology and earlier methods that were more controversial, but also newer analytical techniques that can be used in human forensic DNA identification.
Relevant Expertise
Human DNA identification can involve testimony about laboratory findings, about the statistical interpretation of those findings, and about the underlying principles of molecular biology and genetics. Consequently, expertise in several fields might be required to establish the admissibility of the evidence or to explain it adequately to the trier of fact. The expert who is qualified to testify about laboratory techniques might not be qualified to testify about molecular biology, to make estimates of population frequencies, or to establish that an estimation procedure is valid.12
Trial judges ordinarily are accorded great discretion in deciding when a witness is qualified to testify as an expert, and these decisions depend on the
a final conviction based on substantial evidence against him, (3) he had no new evidence of innocence (only the hope that more extensive DNA testing than that done before the trial would exonerate him), and (4) even a finding that he was not the source of the DNA would not conclusively demonstrate his innocence); Skinner v. Switzer, 562 U.S. 521 (2011); Nat’l Comm’n on the Future of DNA Evidence, Postconviction DNA Testing: Recommendations for Handling Requests (1999); Brandon L. Garrett, Judging Innocence, 108 Colum. L. Rev. 55 (2008); Brandon L. Garrett, Claiming Innocence, 92 Minn. L. Rev. 1629 (2008).
11. See, e.g., Veronica Valdivieso, Note, DNA Warrants: A Panacea for Old, Cold Rape Cases? 90 Geo. L.J. 1009 (2002).
12. See David H. Kaye & Hal S. Stern, Reference Guide on Statistics and Research Methods, section titled “Relevant Expertise,” in this manual. Nonetheless, if previous cases establish that the testing and estimation procedures are legally acceptable, and if the computations are essentially mechanical, then highly specialized statistical expertise might not be essential. Reasonable estimates of DNA characteristics in major population groups can be obtained from standard references, and many quantitatively literate experts could use the appropriate formulae to compute the relevant profile frequencies or probabilities. NRC II, supra note 7, at 170. Limitations in the knowledge of a technician who applies a generally accepted statistical procedure can be explored on cross-examination. E.g., Roberson v. State, 16 S.W.3d 156, 168 (Tex. Crim. App. 2000).
background of each witness. Courts have noted the lack of familiarity of academic experts—who have done respected work in other fields—with the scientific literature on forensic DNA typing and on the extent to which their research or teaching lies in other areas.13 Although such concerns may affect the persuasiveness of particular testimony, they rarely result in exclusion on the grounds that the witness simply is not qualified as an expert on a particular topic.14
Because forensic DNA analysis generally draws on methods and technology developed for research and applications in biological and medical science, the scientific literature on the underpinnings of most forms of DNA evidence, both established and emerging, tends to be extensive. By studying the scientific publications, or perhaps by appointing a special master or expert adviser to assimilate this material,15 a court can ascertain where a party’s expert falls within the spectrum of scientific opinion. Furthermore, an expert appointed by the court under Federal Rule of Evidence 706 could testify about the scientific literature generally or even about the strengths or weaknesses of the particular arguments advanced by the parties.16
Given the diversity of forensic questions to which DNA testing might be applied, it is not feasible to list the specific scientific expertise appropriate to all applications. Some applications span many fields. Consider the range of knowledge required to assess the value of DNA analyses of a novel application such as
13. E.g., State v. Copeland, 922 P.2d 1304, 1318 n.5 (Wash. 1996) (noting that defendant’s statistical expert “was also unfamiliar with publications in the area,” including studies by “a leading expert in the field” whom he thought was “a ‘guy in a lab somewhere’”).
14. E.g., United States v. Pritchard, 993 F. Supp. 2d 1203, 1208–09 (C.D. Cal. 2014) (laboratory analyst “easily meets the standard for qualification” regarding “statistical analysis testimony” because of her academic education and laboratory and courtroom experience, as supplemented by “special training in statistics” at three or four short courses between 8 and 24 hours in length). But see Allen v. State, 62 So. 3d 1199 (Fla. Dist. Ct. App. 2011) (remanding for “a limited evidentiary hearing on the qualifications of the state’s expert to testify as to the statistical significance of the DNA profile matches” where witness had taken statistics in college and received on-site training, but the state did not meet its burden of showing the analyst’s proficiency and general acceptance of the statistical methodology); Gibson v. State, 915 So. 2d 199 (Fla. Dist. Ct. App. 2005) (although the analyst had taken courses in statistics and had testified that she was following the procedure in the 1996 NRC report, the court of appeals “remanded for a limited evidentiary hearing to determine whether the expert had sufficient knowledge of the authoritative sources to present the statistical evidence”). More case law is collected in George L. Blum, Annotation, Qualification as Expert to Testify as to Findings or Results of Scientific Test Concerning DNA Matching, 38 A.L.R. 6th 439 (2008).
15. See, e.g., Gen. Elec. Co. v. Joiner, 522 U.S. 136, 149–50 (1997) (Breyer, J., concurring) (discussing the use of “special masters and specially trained law clerks” in science-related cases); Ass’n of Mex.-Am. Educators v. State of Cal., 231 F.3d 572, 590 (9th Cir. 2000) (“[i]n those rare cases in which outside technical expertise would be helpful to a district court, the court may appoint a technical advisor”); United States v. Lewis, 442 F. Supp. 3d 1122 (D. Minn. 2020) (relying on the report of a special master on the validation and performance of “probabilistic genotyping” software used for the analysis of complex DNA mixtures).
16. See generally Kaye et al., supra note 5, § 12.2; Faigman et al., supra note 5, §§ 1:37 to 1:39.
whole genome sequencing (see section titled “Sequencing and SNPs” below) to establish that a semen sample in a rape case originated from one identical twin rather than the other (see section titled “Twins” below). Expertise in molecular genetics, embryology, biotechnology, bioinformatics, and statistics might be necessary to evaluate the premises and execution of the analysis. Generally, when samples come from crime scenes, the expertise and experience of forensic scientists can be crucial. Just as highly focused specialists may be unaware of aspects of an application outside their field of expertise, so too scientists who have not previously dealt with forensic samples can be unaware of case-specific factors that can confound the interpretation of test results.
Variation in Human DNA and Its Detection
What Are DNA, Chromosomes, Genes, Proteins, and RNAs?
The DNA Molecule
DNA, RNA, and protein molecules are all polymers—large molecules composed of a chain of subunits. The subunits of DNA include four chemical structures known as nucleotide bases. Their names—adenine, thymine, guanine, and cytosine—usually are abbreviated as A, T, G, and C, respectively. The physical structure of DNA is often described as a double helix because the molecule has two spiraling strands connected to each other by weak bonds between the nucleotide bases. As shown in Figure 1, A pairs only with T, and G pairs only with C. Thus, the order of the single bases on either strand reveals the order of the pairs from one end of the molecule to the other, and the DNA molecule represents a long sequence of As, Ts, Gs, and Cs.
Chromosomes
Most human DNA is tightly packed into structures known as chromosomes, which come in different sizes and are located in the nuclei of cells. The chromosomes are numbered, in descending order of size, 1 through 22, with the remaining chromosome being an X or a much smaller Y. Because one of each of these 23 chromosomes is inherited from each parent, the cell’s nucleus normally houses 46 chromosomes in all (see the section titled “Sexual Reproduction and the Genome” below). If the bases are like letters, then each chromosome is like a book written in this four-letter alphabet, and the nucleus is like a bookshelf in the

interior of the cell. All the cells in one individual contain identical copies of the same collection of books.17 The sequence of the As, Ts, Gs, and Cs that constitutes the “text” in these chromosomes is referred to as the individual’s nuclear genome.
All told, the genome has more than three billion “letters” (As, Ts, Gs, and Cs). If these letters were printed in books, the resulting pile would be as high as the Washington Monument. About 99.9% of the genome is identical between any two individuals. This similarity is not really surprising—it accounts for the common features that make modern humans an identifiable species (and for features that we share with many other species as well). The remaining 0.1% is particular to an individual. This variation makes each person (other than identical twins) genetically unique. This small percentage may not sound like a lot, but it adds up to more than three million sites for variation among individuals.
Genes and Gene Products
What are genes, proteins, and RNAs?
Variations in the sequence of base pairs within human populations occur both within the genes and in the regions between the genes. A gene can be defined as
17. Occasional mutations occur, usually as a result of mistakes in the duplication of chromosomes during cell division. See infra note 25.
a segment of DNA, usually from 1,000 to 10,000 base pairs long, that “encodes” a protein or an RNA molecule. For example, a large gene named GC (for group-specific component) encodes a protein that binds to vitamin D and is found in blood plasma. In many people, the following tiny sequence appears within that gene:
G C A A A A T T G C C T G A T G C C A C A C C C A A G G A A C T G G C A.
Unlike double-stranded DNA, RNA is a single helical strand that has a different nucleotide (U, which is short for uracil) in place of the T in the DNA helix. As indicated below, RNA plays an integral part in gene expression.
Proteins are composed of units called amino acids. Twenty different amino acids exist in proteins, and hundreds to thousands of them are attached to each other in long chains with more complex shapes than the single helix of RNA or the double helix of DNA. Proteins perform all sorts of functions in the body and thus produce observable characteristics. For example, the group-specific component protein referred to above (and designated Gc) grabs onto vitamin D and circulates it in the body. It also plays a role in the immune system.
The order of the building blocks of this protein can be slightly different in different individuals. Forensic scientists used these differences before DNA testing was available to exclude or include suspects or defendants as possible sources of blood or semen stains. The cell produces specific proteins and RNAs that correspond to the order of the bases (the “letters”) in the coding part (exons) of a gene. The sequence in which the protein’s amino acids are arranged corresponds to the sequence of base pairs within a gene. A sequence of three base pairs (a codon) specifies a particular 1 of the 20 possible amino acids in the protein. The mapping of various sequences of three nucleotide bases to a particular amino acid is the genetic code. About 1.5% of the human genome codes for the amino-acid sequences.
Human genes also contain noncoding sequences that regulate the cell type in which a protein will be synthesized and how much protein will be produced. Many genes contain interspersed noncoding, nonregulatory sequences that no longer participate in protein synthesis. These sequences, called introns, constitute about 23% of the base pairs within human genes. In terms of the metaphor of DNA as text, the gene is like an important paragraph in the book, often with some gibberish in it.18
18. The idea of a gene as a block of DNA (some of which is coding, some of which is regulatory, and some of which is functionless) is an oversimplification (see, e.g., Petter Portin & Adam Wilkins, The Evolving Definition of the Term “Gene,” 205 Genetics 1353 (2017)), but it is useful enough here.
How do cells manufacture proteins and RNAs?
Protein-coding genes express their sequence-specific information by an intricate process.19 Through a series of biochemical reactions, the base pairs of the exons of the gene are transcribed into an RNA molecule. The noncoding parts (the introns) are not represented in this messenger RNA (mRNA) transcript. The transcribed mRNA makes its way to microscopic molecular factories, where it is translated into a protein.20 By combining the amino acids in different orders, a virtually unlimited variety of proteins can be constructed. Other genes contain DNA sequences that are transcribed into RNAs that perform other functions. Their final products are nonprotein-coding RNAs (ncRNAs).21
Alleles and Loci of Genes
The GC gene mentioned above always is located at the same position, on chromosome 4. But the short sequence listed as occurring within that gene is not the same for every individual. A locus where almost all humans have the same DNA sequence is called monomorphic (“of one form”). A locus where the DNA sequence varies among significant numbers of individuals—more than 1% or so of the population possesses the variant—is called polymorphic (“of many forms”). The alternative forms are called alleles. The GC locus, for example, is
19. The description here of how genes express proteins and RNAs is adapted, in part, from a more complete explanation in the Brief of Genetics, Genomics and Forensic Science Researchers as Amici Curiae in Support of Neither Party in Maryland v. King, 569 U.S. 435 (2013) (reprinted in Henry T. Greely & David H. Kaye, A Brief of Genetics, Genomics and Forensic Science Researchers in Maryland v. King, 53 Jurimetrics J. 43 (2013)) [hereinafter Amici Brief].
20. Transfer RNA brings the building blocks of proteins (the amino acids harvested from food) to these structures. There, they are joined in the order dictated by the mRNA transcript.
21. The ncRNAs include RNAs that do the work of translation and RNAs involved in regulating expression of dozens or even hundreds of protein-coding genes. Furthermore, much shorter RNAs regulate transcription and translation. Thus, it is widely recognized that the genome is abuzz with transcription-to-RNA activity and other events that interact in the expression of the protein-coding DNA. The discoveries of these processes generated speculation in the press and some judicial opinions that all DNA sequences significantly affect development and health. The King amici maintained that
This is not true—even for sequences that are transcribed. Not every biochemical event along the DNA has a measurable impact on health. First, some short ncRNA transcripts are just “noise.” They are degraded quickly. Second, intronic parts of the pre-mRNA transcripts normally are removed. Third, even if one transcript could regulate expression, other transcripts may do the same job. Fourth, even if the stable transcript does affect some trait, the effect may be so minor in the context of other genetic and environmental influences as to be of no meaningful predictive or diagnostic value. Finally, even if a stable transcript has a dramatic effect on a trait, that trait may be unrelated to disease status. Consequently, regarding every bit of the genome as if it were a medical record is unwarranted. Amici Brief, supra note 19, at 11.
polymorphic. The sequence has three common alleles that result from substitutions in a base at a given point. Where an A appears in one allele, there is a C in another. The third allele has the A, but at another point a G is swapped for a T. These changes are called single nucleotide polymorphisms (SNPs, pronounced “snips”).
If a gene is like a paragraph in a book, a SNP is a change in a letter somewhere within that paragraph (a substitution, a deletion, or an insertion), and the two versions of the gene that result from this slight change are the alleles. An individual who inherits the same allele from both parents is called a homozygote. An individual with distinct alleles is a heterozygote.
DNA sequences used for forensic analysis usually are not inside the coding parts of genes. They lie in the vast regions between genes (about 75% of the genome is extragenic) or in the introns. These extra- and intragenic regions of DNA have been found to contain considerable sequence variation, which makes them particularly useful in distinguishing individuals. Although the terms “locus,” “allele,” “homozygous,” and “heterozygous” were developed to describe genes, the nomenclature has been carried over to describe all DNA variation—coding and noncoding alike. Both types are inherited from mother and father in the same fashion, as discussed in the next subsection.
Sexual Reproduction and the Genome
The process that gives rise to the diversity of alleles starts with the production of special sex cells—sperm cells in males and egg cells in females. All the nucleated cells in the body other than sperm and egg cells contain two versions of each of the 23 chromosomes—two copies of chromosome 1, two copies of chromosome 2, and so on, for a total of 46 chromosomes. The 22 numbered chromosomes are called autosomes. Cells in females contain two X chromosomes, and cells in males contain one X and one Y chromosome.22 An egg cell, however, contains only 23 chromosomes—one chromosome 1, one chromosome 2, . . . , one chromosome 22, and one X chromosome—each selected at random from the woman’s full complement of 23 chromosome pairs. Thus, each egg carries half the genetic information present in the mother’s 23 chromosome pairs—a “haploid” genome. Because the assortment of the chromosomes is random, each egg carries a different complement of genetic information. Further randomness comes from a process known as crossing over, in which segments of each of the mother’s two paired chromosomes exchange places in producing the single chromosome from the pair that ends up in
22. A small fraction of people are born with extra chromosomes or a missing one. The most common such condition is Down syndrome (an extra chromosome 21), which occurs in about 1 in every 700 babies born. Nat’l Center on Birth Defects & Developmental Disabilities, Centers for Disease Control and Prevention, Data and Statistics on Down Syndrome (Dec. 16, 2022), https://perma.cc/VB5W-XM5D.
the egg. The same situation exists with sperm cells. Each sperm cell contains a single copy of each of the 23 chromosomes (with crossing over) selected at random from a man’s 23 pairs.23 Fertilization of an egg by a sperm therefore restores the full number of 46 chromosomes, with the 46 chromosomes in the fertilized egg—a “diploid” genome—that is a new combination of those in the mother and father. This recombination of genetic information in sexual reproduction is the main source of human genetic diversity.
During pregnancy, the fertilized cell divides to form two cells, each of which has an identical copy of the 46 chromosomes. The two then divide to form four, the four form eight, and so on. As gestation proceeds, various cells specialize (differentiate) to form different tissues and organs. Although cell differentiation yields many different kinds of cells, the process of cell division usually results in a line of cells having the same genomic complement as the cell that divided. Thus, aside from occasional mutations occurring after fertilization,24 each of the approximately 100 trillion cells in the adult human body has the same DNA as was present in the original 23 pairs of chromosomes from the fertilized egg, one member of each pair having come from the mother and one from the father.25
23. The man’s two sex chromosomes (an X and a Y) are so different that they do not undergo crossing over during the formation of a sperm cell (except in small “pseudo-autosomal regions”—regions at one or both ends of the pair).
24. Mutations in a parent’s sex cell (an egg or sperm) that occur before conception will be incorporated into the genome of the progeny by this repeated copying process. Such prefertilization mutations could complicate parentage or other relationship testing, but they do not matter when testing to see whether two samples originated from the same progeny—for example, a suspect in a criminal case.
25. Mutations can occur during embryonic development or later. The most important source of these changes is a mistake in the DNA copying process during cell division. These are low probability events, but given the immense number of cell divisions that occur during the life of an organism, some are to be expected. Thus, every human being may well be a genetic mosaic to some degree. The mutation rate seems to vary across sites within the genome and between different cell types. Some mutations result in cancers—cells whose proliferation is not inhibited by normal mechanisms. Cristian Tomasetti et al., Stem Cell Divisions, Somatic Mutations, Cancer Etiology, and Cancer Prevention, 355 Science 1330 (2017), https://doi.org/10.1126/science.aaf9011. New cell lineages arising after birth (cancerous or otherwise) do not normally confuse forensic DNA comparisons because the vast majority of them would not occur at the loci used in identity testing (see section titled “What Are DNA Polymorphisms and How Are They Detected?” below), and even if they did, the fraction of the mutated cells in most forensic DNA samples would be so small that only the unmutated alleles would be detected. It is also possible that a mutation would occur “so early in embryonic development that DNA in eggs or sperm might differ from that in blood [or saliva] from the same person.” NRC II, supra note 7, at 64 n.7. The NRC report characterizes this mosaicism as “a remote possibility.” Id. Whether or not this is accurate, as with mutations occurring later in life, the embryonic mutations resulting in the divergence between tissues would have to change the specific loci used in forensic identity testing, which is additionally improbable.
What Are DNA Polymorphisms and How Are They Detected?
By determining which alleles are present at strategically chosen locations on a chromosome (loci), the forensic scientist ascertains the DNA profile, or genotype, of an individual (at those loci). Although the differences among the alleles arise from alternations in the order of nucleotide base pairs (the A, T, G, C letters), DNA typing for ascertaining identity does not require “reading” the full genome. Here we outline the major types of polymorphisms that have been used in identity testing and the methods for detecting them.
VNTRs and RFLP Testing
The first polymorphisms to find widespread use in identity testing result from the presence of a variable number of tandem repeats (VNTRs) at a locus. Being the subject of most of the court opinions on the admissibility of DNA evidence in the late 1980s and early 1990s, they paved the way for the methods now in use. These VNTRs are one type of repetitive DNA. They are like a musical score in which the same melody is played over and over without interruption. The core unit (the melody) is a particular short DNA sequence, and the number of times it is repeated varies from one person to another. The first VNTRs to be used in genetic and forensic testing had core repeat sequences of 7–35 or more base pairs.
In forensic VNTR testing, enzymes (known as restriction enzymes) were used to cut the DNA molecule both before and after the VNTR sequence. A small number of repeats in the VNTR region gives rise to a small restriction fragment, and a large number of repeats yields a large fragment. A substantial quantity of DNA is required to give a detectable number of VNTR fragments with this procedure. After the restriction fragments are sorted by size with a process known as gel electrophoresis,26 the fragments with particular VNTRs are detected by applying a “probe” that binds when it encounters the repeated core sequence. A probe is a short, synthesized fragment of single-stranded DNA with base pairs arranged to complement the core sequence. For example, if the core’s sequence is AGTTC-GCGTGACTAT, the probe’s sequence would be TCAAGCGCACGATA. Because A bonds to T and G to C (and vice versa; see Figure 1), when the probe comes in contact with the restriction fragment, it sticks to it. A radioactive
26. Electrophoresis separates DNA, RNA, or protein molecules according to their size. An electric current drags the molecules through a gel or other matrix. Smaller molecules move through the matrix more quickly than larger molecules. Molecules of known sizes (“standards”) are run through the gel at the same time as the molecules from the sample. Comparing their positions at the end of the run to those of the separated molecules indicates the sizes of the latter.
or fluorescent molecule attached to the probe provides a way to mark the VNTR fragment. Many court opinions refer to this process as RFLP testing.27
PCR Amplification
Most modern forensic-science methods of DNA analysis take advantage of a chemical reaction called PCR (for polymerase chain reaction). The PCR process enables laboratories to make many copies of small DNA fragments.28 Other procedures then can be applied to analyze the vastly amplified quantity of DNA.
PCR can be applied to the double-stranded DNA segments extracted and purified from a forensic sample as follows: First, the purified DNA is separated into two strands by heating it to near the boiling point of water.29 Second, the single strands are cooled, and primers—synthesized fragments of single-stranded DNA, usually between 15 and 30 nucleotides long—attach themselves to the points at which the copying will start and stop.30 Finally, the soup containing the annealed DNA strands, an enzyme called DNA polymerase, and lots of the four nucleotide building blocks are warmed. On a DNA strand, the polymerase inserts the complementary bases one at a time, building a new strand bound to the original template and thus replicating part of the DNA strand that was separated from its partner in the first step. The same replication occurs with the separated partner as the template.31 The result is two identical double-stranded DNA segments, one made from each strand of the original DNA. The three-step cycle is repeated, usually 20 to 35 times in automated machines known as thermal cyclers. Ideally, starting with a single double-stranded molecule, the first cycle results in two double-stranded DNA segments; the second cycle produces four; the third, eight; and so on, until there are millions of copies of the DNA sequences of interest.32
Care must be taken to achieve the appropriate chemical conditions. Furthermore, because even small amounts of stray DNA could be amplified along with the DNA of interest, quality-assurance steps must be taken to reduce contamination
27. It would be clearer to call it RFLP-VNTR testing, because the fragments being measured contain the VNTRs rather than some simpler polymorphisms that were used in genetic research and disease testing. A more detailed exposition of the steps in RFLP-VNTR profiling (including gel electrophoresis, Southern blotting, and autoradiography) can be found in the first edition of this guide (Reference Manual on Scientific Evidence (1st ed. 1994)) and in many judicial opinions circa 1990.
28. Most VNTRs are too long for PCR to work efficiently.
29. This “denaturing” takes about a minute. Chemical and other physical methods for denaturing also can be used.
30. By judiciously constructing the primers to bracket the sequences of interest, the amplified DNA will consist almost entirely of the in-between sequences. The rest of the genome will not be copied. Annealing the primers takes about 45 seconds.
31. This extension step for both templates takes about two minutes.
32. In practice, there is some inefficiency in the doubling process, but the yield from a 30-cycle amplification is generally about 1 million to 10 million copies. NRC II, supra note 7, at 69–70.
of the sample.33 A laboratory should be able to demonstrate that it can amplify targeted sequences faithfully with the equipment and reagents that it uses and that it has taken suitable precautions to minimize or detect contamination from extraneous DNA.34 With small samples, it is possible that some alleles will be amplified and others missed (preferential amplification, discussed below in the section titled “Did the Sample Contain Enough DNA?”). Mutations within a population giving rise to variants in the region of a primer can prevent the amplification of the allele downstream of the primer, giving rise to null alleles.35
STRs and Capillary Electrophoresis
How STRs differ from VNTRs
Although RFLP-VNTR profiling is highly discriminating,36 it has several drawbacks. Not only does it require a substantial sample of DNA-bearing material, but it is time-consuming and does not measure the fragment lengths to the nearest number of repeats.37 Consequently, forensic scientists moved from VNTRs to another form of repetitive DNA known as short tandem repeats
33. In some instances, interpretation of results may be valuable notwithstanding known contaminating DNA. For example, it may be possible to exclude an individual as a viable source. See Human Forensic Biology Subcomm., Organization of Scientific Area Committees for Forensic Science, Standard for Interpreting, Comparing and Reporting DNA Test Results Associated with Failed Controls and Contamination Events, June 1, 2021 (OSAC 2020-S-0004).
34. Forensic-science organizations have proposed guidelines. See, e.g., Eur. Network Forensic Sci. Inst., Guideline for DNA Contamination Minimization in DNA Laboratories, May 10, 2023, https://perma.cc/EQB4-55UB. As have researchers and organizations concerned with PCR-based testing in clinical and research laboratories. E.g., World Health Organization, Dos and Don’ts for Molecular Testing (Jan. 31, 2018), https://perma.cc/LK5B-NBC3.
35. In the context of STR profiling (discussed in the next section), a “null allele is any allele at a microsatellite [STR] locus that consistently fails to amplify to detected levels via the polymerase chain reaction.” Elizabeth E. Dakin & John C. Avise, Microsatellite Null Alleles in Parentage Analysis, 93 Heredity 504, 504 (2004), https://doi.org/10.1038/sj.hdy.6800545. Such a null allele will not lead to a false exclusion if the two DNA samples from the same individual are amplified with the same primer system, but it could lead to an exclusion at one locus when searching a database of STR profiles if the database profile was determined with a different PCR kit than the one used for the crime-scene DNA.
36. Alleles at VNTR loci generally are too long to be measured precisely by electrophoretic methods—alleles differing in size by only a few repeat units may not be distinguished. Although this makes for complications in deciding whether two length measurements that are close together result from the same allele, these loci are quite powerful for the genetic differentiation of individuals, because they tend to have many alleles that occur relatively rarely in the population. At a locus with only 20 such alleles (and most loci typically have many more), there are 210 possible genotypes. With 5 such loci, the number of possible genotypes is 210, which is more than 400 billion.
37. The measurement error inherent in the form of electrophoresis used is not a fundamental obstacle, but it complicates the determination of which profiles match and how often other profiles in the population would be declared to match. For a case reversing a conviction as a result of an
(STRs). STRs have very short core repeats, two to seven base pairs in length, and they typically extend for only some 50 to 350 base pairs.38 Like the larger VNTRs, which extend for thousands of base pairs, STR sequences do not code for proteins or RNAs, and the ones routinely used in identity testing are four-nucleotide repeats thought to have little or no clinical value in ascertaining an individual’s disease status, propensity, or behavioral characteristics.39 The STR loci used in forensic identification have names such as TPOX40 and D16S539. Figure 2 illustrates the nature of allelic variation at the latter locus, on chromosome 16.41 There are other forensic STR loci with more complicated internal patterns.42 Although there are fewer alleles per locus for STRs than for VNTRs, many more STRs are analyzed simultaneously (see section titled “Capillary electrophoresis and fluorescence” below).
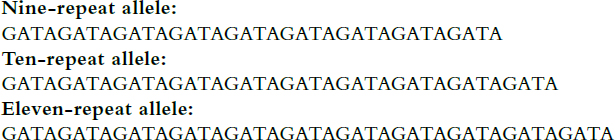 Note: The core sequence is GATA. The first allele listed has nine tandem repeats, the second has ten, and the third has eleven. The locus has other alleles (different numbers of repeats), shown in Figure 4.
Note: The core sequence is GATA. The first allele listed has nine tandem repeats, the second has ten, and the third has eleven. The locus has other alleles (different numbers of repeats), shown in Figure 4.expert’s confusion on this score, see People v. Venegas, 954 P.2d 525 (Cal. 1998). More suitable procedures for match windows and probabilities are described in NRC II, supra note 7.
38. The numbers, and the distinction between “minisatellites” (VNTRs) and “microsatellites” (STRs), are not precise, but the mechanisms that give rise to the shorter tandem repeats differ from those that produce the longer ones. See Jocelyn E. Krebs et al., Lewin’s Genes XII (2018).
39. See Amici Brief, supra note 19; Sara H. Katsanis & Jennifer K. Wagner, Characterization of the Standard and Recommended CODIS Markers, 58 J. Forensic Sci. S169 (2013), https://doi.org/10.1111/j.1556-4029.2012.02253.x; David H. Kaye, Please, Let’s Bury the Junk: The CODIS Loci and the Revelation of Private Information, 102 Nw. U. L. Rev. Colloquy 70 (2007), available at https://perma.cc/8QA3-Y3ES. But see Mayra M. Bañuelos et al., Associations Between Forensic Loci and Expression Levels of Neighboring Genes May Compromise Medical Privacy, 119 Proc. Nat’l Acad. Sci. e2121024119 (2022), https://doi.org/10.1073/pnas.2121024119; Nicole Wyner et al., Forensic Autosomal Short Tandem Repeats and Their Potential Association with Phenotype, 11 Frontiers Genetics 884 (2020), https://doi.org/10.3389/fgene.2020.00884 (concluding that no “forensic STRs . . . were found to be independently causative or predictive of disease,” but proposing that “there remains a strong chance that this inference may change in the near future”).
40. This STR is found in an intron of the thyroid peroxidase gene.
41. Names of the STRs not found in genes are in the form D#S#. The first number indicates the chromosome; the second, a location on the chromosome.
42. For examples, see Peter Gill et al., Forensic Practitioner’s Guide to the Interpretation of Complex DNA Profiles 2–3 (2020).
Medical and human geneticists were interested in VNTRs and STRs as markers in family studies to locate the genes that are associated with inherited diseases. Papers on the potential for identity testing appeared in the early 1990s. Developmental research to pick suitable loci moved into high gear in England and other parts of Europe. Britain’s Forensic Science Service applied a four-locus testing system in 1994. Then it introduced the second-generation multiplex (SGM)—for simultaneously typing six loci in 1996. These soon would be used to build the U.K.’s National DNA Database. The database system allows a computer to check the STR types of millions of known or suspected criminals against thousands of crime-scene samples. A six-locus STR profile can be represented as a string of 12 digits; each digit indicates the number of repeat units in the alleles at each locus. These discrete, numerical DNA profiles are far easier to compare mechanically than the complex patterns of fingerprints. In the United States, the FBI settled on 13 “core loci” to use in the U.S. national DNA database system.43 This number is capable of distinguishing among almost everyone in the population.44 These are often called the CODIS core loci, and an additional seven STR loci were added to the system in 2017.45 The remainder of this section describes how laboratories typically determine which STR alleles are present after DNA from the sample is isolated.
Capillary electrophoresis and fluorescence
Determining which STR alleles are present involves ascertaining the size of the fragments that have been amplified at each STR locus. Separation according to fragment length is done in automated “genetic analyzer” machinery—a byproduct of the technology developed for the Human Genome Project that first sequenced most of the entire genome. In these machines, a long, narrow tube is filled with an entangled polymer or comparable sieving medium, and an electric
43. The federally managed Combined DNA Index System (CODIS) and related issues are discussed in the section titled “Offender and Suspect Database Searches” below; see also John M. Butler, Advanced Topics in Forensic DNA Typing: Methodology 21370 (2012).
44. Usually, there are between 7 and 15 STR alleles per locus. Thirteen loci that have 10 STR alleles each can give rise to 55, or 42 billion trillion, possible genotypes—far, far greater than the U.S. population of 330 million or so. The enormous number of possible 13-locus profiles suggests that it is improbable—but not necessarily impossible—for two unrelated individuals in the country to have identical ones. Even first-degree relatives are unlikely to have the same alleles at all 13 loci. See section titled “Inference, Statistics, and Population Genetics in Human Nuclear DNA Testing” below. But identical twins are expected to share the same STR profile. See section titled “Twins” below.
45. Douglas R. Hares, Selection and Implementation of Expanded CODIS Core Loci in the United States, 17 Forensic Sci. Int’l: Genetics 33 (2015), https://doi.org/10.1016/j.fsigen.2015.03.006. CODIS stands for “combined DNA index system” (discussed in section titled “Offender and Suspect Database Searches” below).
field is applied to pull DNA fragments placed at one end of the tube through the medium. As with gel electrophoresis of RFLPs (see section titled “VNTRs and RFLP Testing” above), shorter fragments slip through the medium more quickly than larger, bulkier ones.
Detecting the fragments as they pass through the capillary tubes is made possible by attaching a fluorescent dye molecule to the PCR primer. As the amplified fragments move through the medium, a laser beam is sent through a small glass window in the tube, causing the dye to glow at a characteristic wavelength. The intensity of the fluorescence is recorded by a kind of electronic camera and transformed into a graph (an electropherogram), which shows a peak as the fragments with the fluorescing dye flash by. Copies of a shorter allele will pass by the window and fluoresce first; copies of a longer fragment will come by later, giving rise to another peak on the graph. Figure 3 is a sketch of how the alleles with five and eight repeats of the GATA sequence at the D16S539 STR locus might appear in an electropherogram.
 Note: One allele has five repeats of the sequence GATA; the other has eight. Each GATA repeat is depicted as a small rectangle. Although only one copy of each allele (with a fluorescent molecule, or “tag” attached) is shown here, PCR generates a great many copies from the DNA sample with these alleles at the D16S539 locus. These copies are drawn through the capillary tube, and the tags glow as the STR fragments move through the laser beam. An electronic camera measures the colored light from the tags. Finally, a computer processes the signal from the camera to produce the electropherogram.
Note: One allele has five repeats of the sequence GATA; the other has eight. Each GATA repeat is depicted as a small rectangle. Although only one copy of each allele (with a fluorescent molecule, or “tag” attached) is shown here, PCR generates a great many copies from the DNA sample with these alleles at the D16S539 locus. These copies are drawn through the capillary tube, and the tags glow as the STR fragments move through the laser beam. An electronic camera measures the colored light from the tags. Finally, a computer processes the signal from the camera to produce the electropherogram.Source: David H. Kaye, The Double Helix and the Law of Evidence 189, fig. 9.1 (2010).
Modern genetic analyzers produce electropherograms for many loci at once.46 This “multiplexing” is accomplished by using dyes that fluoresce at distinct colors to label the alleles from different groups of loci. A separate set of fragments of known sizes that comigrate through the capillary function as a kind of ruler (an internal-lane size standard) to determine the lengths of the allelic fragments. Software processes the raw data to generate an electropherogram of the separate allele peaks of each color. By comparing the positions of the allele peaks to the size standard, the program determines the number of repeats in each allele. The plotted heights of the peaks (measured in relative fluorescent units, or RFUs) are proportional to the amount of the PCR product.
Complications can arise, especially with minute samples and mixtures. During PCR, the DNA strand being copied and the new strand can slip, causing “stutter” peaks that typically are one repeat away from the originating STR alleles. Fragments of DNA can fall into PCR tubes or be carried by dust particles, causing “drop in” alleles; color dyes do not fluoresce solely at the desired color and can give a signal in the adjacent color channel—a spectral artifact known as pull-up; a variety of factors can lead to unequal peaks for the pair of alleles at a locus (peak imbalance) and to missing peaks (allele dropout or even whole locus dropout).47
Figure 4 is an electropherogram of all 203 major alleles at 15 STR loci typed in a single multiplex PCR reaction. In addition, it shows the two alleles of the gene used to determine the biological sex of the contributor of a DNA sample.48 An electropherogram from an individual’s DNA would have only one or two peaks at each of these 15 STR loci (depending on whether the person is homozygous or heterozygous). These “allelic ladders” aid in deciding which allele a peak from an unknown sample represents.
46. Kathryn Oostdik et al., Developmental Validation of the PowerPlex® Fusion System for Analysis of Casework and Reference Samples: A 24-locus Multiplex for New Database Standards, 12 Forensic Sci. Int’l Genetics 69 (2014), https://doi.org/10.1016/j.fsigen.2014.04.013; Suhua Zhang et al., Development and Validation of a New STR 25-plex Typing System, 17 Forensic Sci. Int’l: Genetics 61 (2015), https://doi.org/10.1016/j.fsigen.2015.03.008.
47. See also sections titled “Did the Sample Contain Enough DNA?” and “Mixtures” below.
48. The amelogenin gene, which is found on the X and the Y chromosome, codes for a protein that is a major component of the tooth enamel matrix. The copy on the Y chromosome is 112 base pairs (bp) long. The copy on the X chromosome has a string of six base pairs deleted, making it slightly shorter (106 bp). A female (XX) will have one peak at 112 bp. A male (XY) will have two peaks (at 106 and 112 bp). In some populations, however, mutations that interfere with the detection of the Y chromosome by this method have been observed. These could lead to male DNA being misidentified as female on the basis of this locus alone. See Vijendra Kumar Kashyap et al., Deletions in the Y-derived Amelogenin Gene Fragment in the Indian Population, 7 BMC Med. Genetics 37 (2006), https://doi.org/10.1186/1471-2350-7-37.
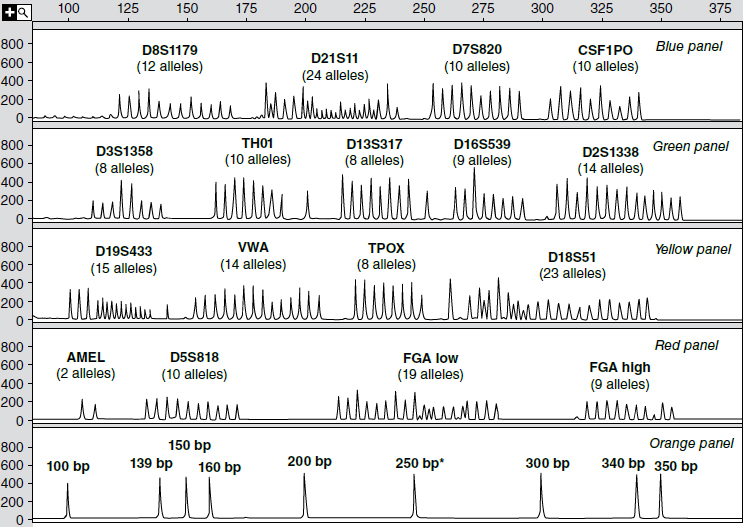 Note: The bottom panel is a sizing standard—a set of peaks from DNA sequences of known lengths (in base pairs). The numbers in the vertical axis in each panel are relative fluorescence units (RFUs) that indicate the amount of light emitted after the laser beam strikes the fluorescent tag on an STR fragment. Applied Biosystems makes the kit that produced these allelic ladders.
Note: The bottom panel is a sizing standard—a set of peaks from DNA sequences of known lengths (in base pairs). The numbers in the vertical axis in each panel are relative fluorescence units (RFUs) that indicate the amount of light emitted after the laser beam strikes the fluorescent tag on an STR fragment. Applied Biosystems makes the kit that produced these allelic ladders.Source: John M. Butler, Forensic DNA Typing: Biology, Technology, and Genetics of STR Markers 128 (2d ed. 2005). Copyright Elsevier 2005, with the permission of Elsevier Academic Press. John Butler supplied the illustration.
Figure 5 is an electropherogram from the vaginal epithelial cells of the body of a girl who had been sexually assaulted and killed in California.49 It was produced for the retrial in 2008 of the defendant, who was linked to the victim by VNTR typing at his first trial in 1990.
49. People v. Pizarro, 12 Cal. Rptr. 2d 436 (Ct. App. 1992), after remand, 3 Cal. Rptr. 3d 21 (Ct. App. 2003), review denied (Oct. 15, 2003).
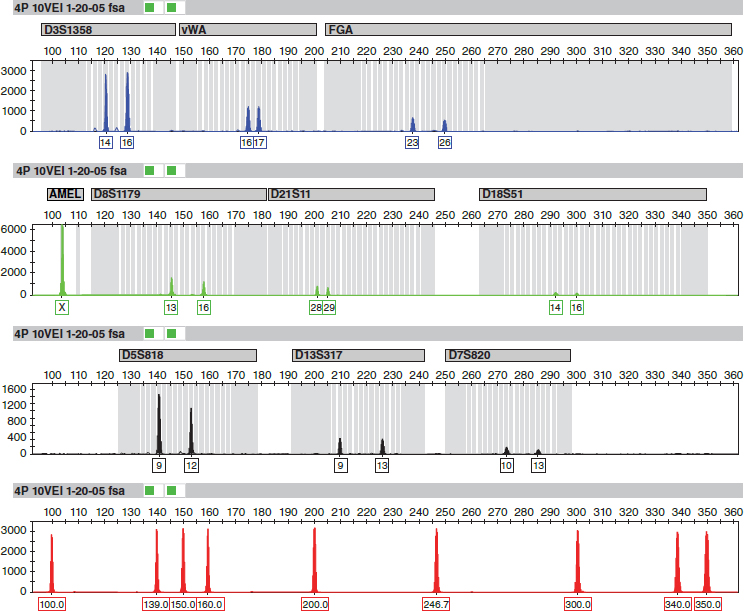 Note: The amelogenin locus and a sizing standard at the bottom also are included. Some STR loci have small peaks, indicating that there was not much PCR product for those loci, likely because of DNA degradation. All of the STR loci have two peaks, as would be expected when the source is heterozygous at those loci.
Note: The amelogenin locus and a sizing standard at the bottom also are included. Some STR loci have small peaks, indicating that there was not much PCR product for those loci, likely because of DNA degradation. All of the STR loci have two peaks, as would be expected when the source is heterozygous at those loci.Source: Steven Myers and Jeanette Wallin, California Department of Justice, provided the image.
Miniaturization for rapid capillary electrophoresis
Miniaturized capillary electrophoresis (CE) devices have been developed for rapid detection of STRs and other genetic analyses. The mini-CE systems consist of microchannels etched on glass, silicon, or plastic wafers (“chips”) using technology borrowed from the computer industry. The microchannels are roughly the diameter of a hair. The principles of electrophoretic separation are the same as with conventional CE systems, but with microfluidic technologies, it is possible to integrate DNA extraction and PCR amplification processes with the CE separation in a single device, a so-called lab on a chip instrument that includes a cartridge with miniature pumps, mixers, valves, fluid channels
and reagent storage chambers.50 Once a sample is added to the cartridge, all the analytical steps are performed without further human contact. These devices combine simplified sample handling with rapid analysis.
“Rapid DNA” devices have been commercialized for point-of-care medical diagnostics51 and for military and police applications. Validation for police work can be achieved by having police obtain profiles under realistic conditions and then rerunning the samples with conventional equipment at established laboratories; or samples from known sources of DNA can be created for testing under experimental conditions.52 Several different rapid DNA machines for forensic STR profiling have been used by police departments or other agencies across the world in various ways: (1) to acquire a DNA profile during an initial encounter with a suspect; (2) to obtain the profile quickly after arrest as part of the booking process; (3) to extract and analyze DNA from crime-scene and sexual-assault-kit samples; and (4) to identify victims of mass disasters.53 In the first situation, police may lack probable cause to arrest an individual but may try to conform to the requirements of the Fourth Amendment by securing valid consent to the DNA profiling.54 Although some local police agencies—and a large prosecutors’ office—have pursued this
50. Lab-on-a-chip instruments (often called LOCs) also can use other methods than CE for analyzing DNA, proteins, or other molecules. See, e.g., Prapti Pattanayak et al., Microfluidic Chips: Recent Advances, Critical Strategies in Design, Applications and Future Perspectives, 25 Microfluidics & Nanofluidics 99 (2021), https://doi.org/10.1007/s10404-021-02502-2.
51. Curtis D. Chin et al., Commercialization of Microfluidic Point-of-Care Diagnostic Devices, 12 Lab on a Chip 2118 (2012), https://doi.org/10.1039/c2lc21204h; P. Yager et al., Microfluidic Diagnostic Technologies for Global Public Health, 442 Nature 412 (2006), https://doi.org/10.1038/nature05064. However, those applications would not use human STRs; instead, they would detect the DNA or RNA of pathogens.
52. E.g., Rosemary S. Turingan et al., Developmental Validation of the ANDE 6C System for Rapid DNA Analysis of Forensic Casework and DVI Samples, 65 J. Forensic Sci. 1056 (2020), https://doi.org/10.1111/1556-4029.14286. More detailed guidelines for validation can be found in the United Kingdom Forensic Science Regulator’s report, Forensic Science Regulator Guidance: Methods Employing Rapid DNA Devices § 8 (2021), https://perma.cc/746P-9XW3 (FSR-G-229).
53. The current systems take about 90 minutes to analyze a sample.
54. Some localities go further and keep the STR data in a computer-searchable, local DNA database that they can operate independently of the databases that are part of the system of shared profile data (known as CODIS), which are administered by the FBI pursuant to the DNA Identification Act of 1994, 34 U.S.C. §§ 40702–40703 (later amended in various respects) and state statutes. Whether the indefinite retention and trawling of the local database of consent profiles for matches to future crime-scene samples exceeds the scope of the consent in a given case could be a significant question. Moreover, the non-CODIS local databases (most of which were created without rapid DNA instruments) have been criticized on other grounds. See, e.g., Jason Kreag, Going Local: The Fragmentation of Genetic Surveillance, 95 B.U. L. Rev. 1491 (2015); N.Y. City Bar Ass’n Crim. Courts Comm., Crim. Just. Operations Comm., and Mass Incarceration Task Force, Curbing Unregulated Local DNA Indexing, Apr. 16, 2021 (report supporting “an Act . . . requiring municipalities to expunge any DNA record stored in a municipal DNA identification index,” accessible via https://perma.cc/72FC-PGL7). In Arizona, the state Department of Public Safety chose to establish a non-CODIS database, relying on rapid DNA instruments and DNA database
practice,55 rapid DNA technology has been more widely adopted as an adjunct to arrests. Pursuant to the Rapid DNA Act of 2017,56 the FBI issued standards and procedures for the use of rapid DNA instruments in the booking station that allow the resulting DNA profiles to be uploaded to the National DNA Index System.57 It is working cautiously toward the third approach, of rapid DNA analysis of crime-scene and sexual-assault-victim DNA samples.58
Rather than face objections to rapid DNA based on Rule 702, prosecutors might choose to have accredited laboratories redo the analyses that police perform and introduce only the latter evidence. This will be possible for fresh DNA samples from defendants, but not when the crime-scene DNA has been totally consumed during the rapid testing. As with the widespread use of Breathalyzers for blood-alcohol testing,59 ultimately courts will confront Rule 702 issues of scientific validity and proper use of the equipment. A number of organizations and individuals have expressed reservations about the current state of the technology,60 and research is ongoing.61
management software purchased from vendors, for the benefit of localities that otherwise might have been tempted to establish such systems on their own. Kreag, supra, at 1517–19.
55. In Orange County, California, prosecutors with rapid DNA machines in their basement assembled a significant database by “offer[ing] defendants accused of misdemeanors and infractions a deal: give the prosecutor’s office your DNA, and the office will offer you leniency in your criminal case.” Andrea Roth, “Spit and Acquit”: Prosecutors as Surveillance Entrepreneurs, 107 Cal. L. Rev. 405, 408 (2019). California Senate Bill 1228 (adopted 2022) curbs such practices.
56. Pub. L. No. 115–50, 131 Stat. 1001 (codified at 34 U.S.C. §§ 12591, 12592, 40702, 40703).
57. See FBI, Guide to All Things Rapid DNA, Jan. 27, 2022, https://le.fbi.gov/file-repository/rapid-dna-guide-january-2022.pdf/view. Previously, only qualified state laboratories could submit profiles.
58. Id.
59. See 1 McCormick on Evidence, supra note 1, § 205.1, at 1309–10 n.12.
60. There are concerns over fully automated analysis of small samples and mixtures because of sample consumption, reduced sensitivity relative to full-scale equipment, and the integrated software for interpretation. Discussions of the state of the art and research and administrative recommendations can be found in, for example, Erik Dalin et al., Biology Section, Nat’l Forensic Centre, Swedish Police Authority, Rapid DNA: A Summary of Available Rapid DNA Systems (2022), https://perma.cc/87XS-WQ9W; Douglas R. Hares et al., Rapid DNA for Crime Scene Use: Enhancements and Data Needed to Consider Use on Forensic Evidence for State and National DNA Databasing—An Agreed Position Statement by ENFSI, SWGDAM and the Rapid DNA Crime Scene Technology Advancement Task Group, 48 Forensic Sci. Int’l: Genetics 102349 (2020), https://doi.org/10.1016/j.fsigen.2020.102349; Non-CODIS Rapid DNA Best Practices/Outreach and Courtroom Considerations Task Group, Texas Forensic Sci. Comm’n, Non-CODIS Rapid DNA Considerations and Best Practices for Law Enforcement Use, Sept. 16, 2019, https://perma.cc/X3VV-RCUB.
61. E.g., Belinda Martin et al., Analysis of Rapid HIT Application to Touch DNA Samples, 67 J. Forensic Sci. 1233 (2022), https://doi.org/10.1111/1556-4029.14964 (“not fit for the routine analysis of touch DNA samples in forensic casework”); Denise Ward et al., Analysis of Mixed DNA Profiles from the RapidHIT™ ID Platform Using Probabilistic Genotyping Software STRmix™, 58 Forensic Sci. Int’l: Genetics 102664 (2022), https://doi.org/10.1016/j.fsigen.2022.102664 (“good discrimination power [when STR data was analyzed with external software] but less than those produced via the standard laboratory workflow,” as would be “expected . . . for the advantages of speed and portability”).
Sequence-Specific Probes and Microarrays
Simple sequence variation, such as that for the GC locus (see section titled “Chromosomes” above), is conveniently detected using sequence-specific probes (defined in the sections on RFLP-VNTR and STR profiling). With GC typing, for example, probes for the three common alleles are attached at three separate spots on a membrane. Copies of the variable sequence region of the GC gene in the crime-scene sample are made with PCR. These copies (in the form of single strands) are poured onto the membrane. Whichever allele is present in a copy will cause it to stick to a corresponding, immobilized probe strand. A chemical “label” that catalyzes a color change at the spot where the trace-evidence allele binds to its probe can be attached when the copies are made. A colored spot showing that the allele is present thus should appear on the membrane at the location of the probes that correspond to this particular allele. If only one allele is present in the crime-scene DNA (because of homozygosity), there will be no color change at the spots where the other probes are located. If two alleles are present (heterozygosity), the corresponding two spots will change color.
This method was used before STR profiling, with only a few loci.62 In that form, it lacked the discriminating power of STRs and VNTRs, but, being PCR-based, it was more sensitive, and easier and quicker to perform than RFLP-VNTR profiling. Admissibility followed.63
The approach can be miniaturized and automated by embedding probes for many loci on a chip.64 Such a “microarray” for DNA-sequence detection usually consists of a two-dimensional grid of many thousands of microscopic spots on a silicon, glass, plastic, or paper surface. Each spot contains many copies of a probe with its own particular sequence, tethered to the surface at one end. A solution containing copies of single-stranded target DNA fragments65 (with attached fluorescent tags or dyes) is washed over the microarray surface. As usual, the probes on the array bind to copies with the complementary sequence. The spots
62. Indeed, in what probably was the first criminal case in the United States in which DNA evidence was admitted, a single-locus test that suggested that the owners of a nursing home, although found guilty of negligent homicide for the starvation of an elderly patient, did not swap the organs of the victim with those of another cadaver in an alleged attempt to cover up the cause of death. Stephen Michaud, DNA Detectives, N.Y. Times Mag., Nov. 6, 1988, § 6, at 70, available at https://www.nytimes.com/1988/11/06/magazine/dna-detectives.html.
63. On the development and judicial reception of the “dot blot” or “reverse dot blot” tests and the “polymarker” system, see Kaye, supra note 1, at 180–87.
64. See, e.g., Johannes Wöhrle et al., Digital DNA Microarray Generation on Glass Substrates, 10 Sci. Reps. 5770 (2020), https://doi.org/10.1038/s41598-020-62404-1.
65. Amplification (including isothermal methods that do not require the heating and cooling cycles of conventional PCR) of the target DNA can be built into the microarray. Roger Bumgarner, Overview of DNA Microarrays: Types, Applications, and Their Future, 101 Current Protocols in Molecular Biology 22–1 (2013), https://doi.org/10.1002/0471142727.mb2201s101.
that capture the targeted DNA are identified, indicating the presence of those sequences in the sample.
DNA microarrays have broad applications in biology and medicine.66 Forensic applications include sequencing human mitochondrial DNA (see section titled “Mitochondrial DNA” below) and resolving individual genotypes within complex DNA mixtures,67 but the most well known application comes from SNP chips with enough different probes to detect half-a-million or more different known SNPs distributed throughout the human genome.68 After biotechnology companies produced these microarrays for genomics research and health applications,69 direct-to-consumer genetic testing companies popularized them for ancestry and other “recreational genetics” activities. As more and more people interested in locating possibly unknown relatives elected to share certain genomic information in a public database, trawling the database for putative relatives of perpetrators of cold cases to infer the identity of the unknown source of the crime-scene DNA proved spectacularly successful. This procedure of investigative genetic genealogy is the subject of a later section by that name.
Sequencing and SNPs
As shown in Figure 1 (see section titled “The DNA Molecule” above), DNA contains a sequence of paired letters—the nucleotides A, T, G, and C—laid out
66. They permit large-scale population studies—for example, to determine how often individuals with a particular mutation develop breast cancer, or to identify the changes in gene sequences that are most often associated with particular diseases. Microarrays also are used in studies of variation in the number of copies of certain genes in different people’s genomes (copy number variation). They are used to study the extent to which certain genes are turned on or off in cells and tissues. For this purpose, instead of isolating DNA from the samples, a transcript of the DNA (mRNA, supra section titled “Chromosomes”) is isolated and measured. They provide clinical diagnostic tests for diseases. “Biosensor” microarrays detect pathogens and other targets.
67. Lev Voskoboinik et al., SNP-Microarrays Can Accurately Identify the Presence of an Individual in Complex Forensic DNA Mixtures, 16 Forensic Sci. Int’l: Genetics 208 (2015), https://doi.org/10.1016/j.fsigen.2015.01.009 (proof of concept).
68. Another proposed use is providing SNP data for better discerning the distinct contributors to DNA mixtures. Nils Homer et al., Resolving Individuals Contributing Trace Amounts of DNA to Highly Complex Mixtures Using High-Density SNP Genotyping Microarrays, 4 PLoS Genetics e1000167 (2008), https://doi.org/10.1371/journal.pgen.1000167. But see Rosemary Braun et al., Needles in the Haystack: Identifying Individuals Present in Pooled Genomic Data, 5 PLoS Genetics e1000668 (2009), https://doi.org/10.1371/journal.pgen.1000668; Peter M. Visscher & William G. Hill, The Limits of Individual Identification from Sample Allele Frequencies: Theory and Statistical Analysis, 5 PLoS Genetics e1000628, https://doi.org/10.1371/journal.pgen.1000628.
69. One study of 3,000 Europeans used a commercial microarray with over half a million SNPs “to infer [the individuals’] geographic origin with surprising accuracy—often to within a few hundred kilometers.” John Novembre et al., Genes Mirror Geography Within Europe, 456 Nature 98, 98 (2008), https://doi.org/10.1038/nature07331.
in a spiraling kind of line. Broadly, sequencing refers to ascertaining the precise order of the DNA letters along each strand.70 The sequence-specific probes of the previous section could be said to sequence short segments of DNA in those instances in which they bind to the complementary sequence in the target DNA. But that is not how DNA sequencing machines work. They determine the order of base pairs one at a time, somewhat like spelling out the letters of a word before one recognizes it as a word. This sequencing is the subject of this section. It can be done for short stretches of DNA or, with enough effort and ingenuity, and in a roundabout way, for the whole genome, ultimately revealing the order of the base pairs at every gene, every STR, and every other kind of locus.
Sequencing methods
Sequencing technology is hardly new. In the 1970s, biochemists developed two PCR-related methods to sequence DNA.71 For the next 30 years, researchers applied “Sanger sequencing” to decipher complete genes and, later, entire genomes of organisms. It is still used to sequence particular regions (up to a thousand or so base pairs). Basically, it uses PCR to synthesize a series of complementary DNA strands with color-coded nucleotides that indicate whether the nucleotide in the template strand is an A, T, G, or C.72 The multinational, multiyear Human Genome Project, completed in the early 2000s, employed factory-like systems with hundreds of sequencing machines to Sanger sequence most of the 3.2 billion base pairs in the haploid genome from several anonymous
70. Because of the strict A-T, G-C pairing rule, it is not necessary to write two letters to designate a pair; the sequence of bases on one strand implies the sequence on the other.
71. Allan M. Maxam & Walter Gilbert, A New Method for Sequencing DNA, 74 Proc. Nat’l. Acad. Sci. 560 (1977), https://doi.org/10.1073/pnas.74.2.560; Frederick Sanger et al., DNA Sequencing with Chain-terminating Inhibitors, 74 Proc. Nat’l. Acad. Sci. 5463 (1977), https://doi.org/10.1073/pnas.74.12.5463.
72. As with ordinary PCR amplification, the target DNA is unzipped and copied—but the polymerase chain reaction stops prematurely because some “chain-terminating” nucleotides (the A, T, C, and G units) are added to the PCR mixture. The chemically modified nucleotides are missing the molecular “hook” that lets the next unit attach itself to the growing chain, and they have a fluorescent dye attached to them (with a different color for A, T, G, and C units). The chain reaction continues adding nucleotides until it happens to add a modified one. This process is repeated many times to generate fragments that extend to all possible stopping points. The smallest ones will consist of only one terminating unit after the primer. The next smallest will have two nucleotides, and so on, for every base pair in the original target DNA fragment. Capillary electrophoresis (supra section titled “STRs and Capillary Electrophoresis”) separates the synthesized fragments from smallest to largest, with the results displayed as colored peaks in the electropherogram (also called a chromatogram). The order of the colored peaks gives the sequence of the base pairs in the target DNA. The whole process is an example of sequencing by synthesis.
individuals.73 Despite the ultimate success, the sequencing efforts were technically demanding, expensive, time-consuming, and not suitable for regions on the chromosomes that are replete with repetitive DNA.74
In 2004, the National Human Genome Research Institute announced funding for research leading to the “$1,000 genome,” which would permit sequencing an individual’s genome for medical diagnosis and improved drug therapies. The goal was essentially met in 2014 thanks to methods known as next-generation sequencing (NGS) or massively parallel sequencing (MPS).75 MPS methods simultaneously read millions of fragments and then use computer-intensive alignment programs to stitch the reads together to give the whole genome sequence. The genomes can be small (as with bacteria), large (as with humans), or larger still (as with some plants and other organisms). The methods also can be applied to selected parts of a genome, making them attractive for forensic and diagnostic purposes.
Commercially available sequencers implement the MPS approach in different ways.76 One popular system is another form of sequencing by synthesis. First, to prepare the sample for sequencing, all the DNA is cut into many little pieces, or the targets are amplified as small pieces, creating a “library” of fragments. Then “adapters” are added to the ends of the pieces in this library. These adapters are like tiny handles that help immobilize the DNA to facilitate sequencing. Second, instead of running PCR in a tube, the DNA is loaded onto a glass “flow cell” that has millions of tiny wells on its surface. Each of these wells can grab an adapter to capture a single piece of DNA from the library. Third, the fragments in the wells are amplified with PCR, resulting in a cluster of identical single-stranded DNA in a given well. Fourth, chemically modified nucleotides bind to the DNA template strand. As in Sanger sequencing, each nucleotide contains a fluorescent tag, but the terminator is removed after the color at each well is detected, allowing the next base to bind. The reactions are repeated hundreds of times to trace the sequence of nucleotides in the growing chain. The cycle of one-pair-at-a-time synthesis occurs in millions of wells at once. Finally, bioinformatics software fits the jigsaw puzzle of millions of pieces together.77
73. Int’l Human Genome Sequencing Consortium, Finishing the Euchromatic Sequence of the Human Genome, 431 Nature 931 (2004), https://doi.org/10.1038/nature03001; Int’l Human Genome Sequencing Consortium: Initial Sequencing and Analysis of the Human Genome, 409 Nature 860 (2001), https://doi.org/10.1038/35057062; cf. J. Craig Venter et al., The Sequence of the Human Genome, 291 Science 1304 (2001), https://doi.org/10.1126/science.1058040.
74. More efficient methods have enabled researchers to fill in the gaps, correct errors, and release “a truly complete human reference genome.” Sergey Nurk et al., The Complete Sequence of a Human Genome, 376 Science 44 (2022), https://doi.org/10.1126/science.abj6987.
75. Erwin L. van Dijk et al., Ten Years of Next-Generation Sequencing Technology, 30 Trends in Genetics 418 (2014), https://doi.org/10.1016/j.tig.2014.07.001.
76. Barton E. Slatko et al., Overview of Next-Generation Sequencing Technologies, 122 Current Protocols in Molecular Biology e59 (2018), https://doi.org/10.1002/cpmb.59.
77. With MPS, each base has to be read not just once, but at least several times in the overlapping segments to ensure accuracy in fitting the reads together.
The MPS sequencing-by-synthesis method described above sometimes is cited as an example of second-generation sequencing. Its reads are much shorter and more prone to error (per read) than Sanger sequencing,78 but the speed and cost are enormous improvements for sequencing large genomes.
The sequencing part of the process (the steps starting with insertion of the library of DNA fragments into the flow cell) need not be applied to whole genomes. The library can consist of copies of short segments derived from any interesting sequences.79 MPS thus supports targeted sequencing of many polymorphisms of forensic interest all at once.80
A second example of an MPS technology has been called third- or even fourth-generation sequencing. It produces long reads from a single DNA molecule without having to amplify it by PCR and to attach and illuminate fluorescent labels. Proteins embedded into a biological membrane create exquisitely small holes, called nanopores. (A human hair is on the order of 100,000 nanometers wide; a DNA molecule is 2–12 nanometers wide.) Either single- or double-stranded DNA can be moved through multiple nanopores of a flow cell at a constant rate for tens of thousands of nucleotides. As a DNA molecule is ratcheted through a pore, it interrupts a flow of electrical current to a sensor chip. The disruption produces a disturbance in the current that is characteristic of the type of nucleotide (A, T, G, or C) passing through. Software decodes the disturbance to determine the DNA sequence as the nucleotides flow by.
With such long-read sequencing, assembling the overlapping reads is easier, just as putting together a jigsaw puzzle with fewer, larger pieces is easier than fitting together numerous smaller ones. Moreover, large insertions or deletions and repetitive regions can be detected more easily. Although the longer reads can have a higher error rate, ongoing technological improvements are closing the accuracy gap with the sequencing-by-synthesis versions of MPS.81
78. Sanger sequencing platforms “offer well-trusted, up to 1-kbp long reads . . . and are still considered the gold standard for sequencing. They are routinely used for clinical gene tests or validations. However, their low-throughput limits them to single or small batches of targets.” Adam Ameur et al., Single-Molecule Sequencing: Towards Clinical Applications, 37 Trends in Biotech. 72 (2019), https://doi.org/10.1016/j.tibtech.2018.07.013.
79. Panels of genes with variants that are known to promote the growth of cancer cells can be targeted to see which mutations are present in cancer patients. See, e.g., Masayuki Nagahashiet et al., Next Generation Sequencing-based Gene Panel Tests for the Management of Solid Tumors, 110 Cancer Sci. 6 (2019), https://doi.org/10.1111/cas.13837.
80. Peter de Knijff, From Next Generation Sequencing to Now Generation Sequencing in Forensics, 38 Forensic Sci. Int’l: Genetics 175 (2019), https://doi.org/10.1016/j.fsigen.2018.10.017.
81. E.g., Søren M. Karst et al., High-Accuracy Long-Read Amplicon Sequences Using Unique Molecular Identifiers with Nanopore or Pacbio Sequencing, 18 Nature Methods 165 (2021), https://doi.org/10.1038/s41592-020-01041-y.
High-throughput sequencing technologies have demonstrated their usefulness in a wide range of research,82 clinical,83 and public health84 applications. A famous example is the whole-genome sequencing of highly degraded ancient DNA (aDNA) of extinct species, including woolly mammoths, cave bears, Neanderthals, and Denisovans, as well as more modern human populations, from Vikings to Paleo-Inuit.85
The range of forensic applications
Of course, different applications within the broad category of massively parallel or next-generation sequencing can use different techniques, and particular forensic applications need to be validated to demonstrate that they are fit for their intended use. Research has shown that MPS can be used for the following:
- to increase the power of STR loci to distinguish among individuals by detecting sequence differences within the length-based alleles as measured by capillary electrophoresis;86
82. E.g., Vivian Marx, Method of the Year: Long-read Sequencing, 20 Nature Methods 6 (2023), https://doi.org/10.1038/s41592-022-01730-w; Katia Nones & Ann-Marie Patch, The Impact of Next Generation Sequencing in Cancer Research, 12 Cancers (Basel) 2928 (2020), https://doi.org/10.3390/cancers12102928.
83. Elaine Hsu et al., Rapid Pathogen Detection by Metagenomic Next-Generation Sequencing of Infected Body Fluids, 27 Nature Med. 115 (2021), https://doi.org/10.1038/s41591-020-1105-z;F. Mosele et al., Recommendations for the Use of Next-Generation Sequencing (NGS) for Patients with Metastatic Cancers: A Report from the ESMO Precision Medicine Working Group, 31 Annals Oncology 1491 (2020), https://doi.org/10.1016/j.annonc.2020.07.014. But see Francois Balloux et al., From Theory to Practice: Translating Whole-Genome Sequencing (WGS) into the Clinic, 26 Trends in Microbiology 1035 (2018), https://doi.org/10.1016/j.tim.2018.08.004 (discussing obstacles to more routine use); Nagahashiet et al., supra note 79.
84. E.g., Rowena A. Bull et al., Analytical Validity of Nanopore Sequencing for Rapid SARSCoV-2 Genome Analysis, 11 Nature Commun. 6272 (2020), https://doi.org/10.1038/s41467-020-20075-6; David F. Nieuwenhuijse et al., Towards Reliable Whole Genome Sequencing for Outbreak Preparedness and Response, 23 BMC Genomics 569 (2022), https://doi.org/10.1186/s12864-022-08749-5; Joshua Quick et al., Real-time, Portable Genome Sequencing for Ebola Surveillance, 530 Nature 228 (2016), https://doi.org/10.1038/nature16996. By sequencing entire bacterial genomes, researchers can rapidly differentiate organisms that have been genetically modified for biological warfare or terrorism from routine clinical and environmental strains. B. La Scola et al., Rapid Comparative Genomic Analysis for Clinical Microbiology: The Francisella Tularensis Paradigm, 18 Genome Res. 742 (2008), https://doi.org/10.1101/gr.071266.107.
85. Elizabeth D. Jones, Ancient DNA: The Making of a Celebrity Science (2022); Andrew Curry, Ancient DNA Pioneer Svante Pääbo Wins Nobel, 378 Science 12 (2022), https://doi.org/10.1126/science.adf1845.
86. For example, one allele might have nine copies of a GATA repeat (as in Figure 2), while another might have eight GATA repeats and one GTTA instead of a GATA. Their PCR products, being the same length, would migrate through the gel at the same speed, so they would show up as a single peak on an electropherogram. So would two alleles—from a different individual—that are
- to better resolve mixtures into the components coming from different contributors (as discussed in the section titled “Mixtures” below);
- to develop investigative leads based on biogeographic ancestry and physical characteristics such as eye color;87
- to ascertain the tissues that the DNA in crime-scene samples came from when the whole cells are not present in the samples;88
- to sequence mitochondrial DNA;89 and even
- to distinguish between identical twins.90
Moreover, as explained in the next section, the ability of MPS to find point mutations (insertions, deletions, or substitutions of single base pairs) opens up the possibility of using well-chosen SNPs as a replacement for (or at least a supplement to) STR profiling for the purpose of individual identification, that is, of determining whose DNA is in a crime-scene sample.91 Biotechnology companies have developed kits for preparing the libraries for all these purposes,92 and in 2024, MPS results were held to be admissible for the first time in the United States.93
SNPs as identification loci
Single-nucleotide polymorphisms (SNPs) are the most abundant variations in the human genome. SNPs are shorter than STRs, making it possible to type degraded samples that regular STR profiling cannot handle because the
made up of nine GATA repeats. PCR-CE would not distinguish between these two individuals, but the more detailed sequence information could. The additional information also can improve mixture resolution (see infra section titled “Mixtures”).
87. See infra section titled “Investigative DNA Analysis.”
88. See infra section titled “DNA and RNA Typing of Bodily Fluids or Tissues.”
89. See infra section titled “Mitochondrial DNA.”
90. See infra section titled “Twins.”
91. See, e.g., Christopher P. Phillips et al., A Compilation of Tri-allelic SNPs from 1000 Genomes and Use of the Most Polymorphic Loci for a Large-Scale Human Identification Panel, 46 Forensic Sci. Int’l: Genetics 102232 (2020), https://doi.org/10.1016/j.fsigen.2020.102232.
92. Penelope R. Haddrill. Developments in Forensic DNA Analysis, 5 Emerging Topics Life Sci. 381 (2021), https://doi.org/10.1042/ETLS20200304.
93. People v. Chavez, No. BF187877A (Kern Cnty. Cal. Super. Ct. Jan. 22, 2024). Defendant was convicted of first-degree murders of two homeless women. In this unreported case, “[f]orensic evidence, including DNA and fingerprints, directly linked [the defendant] to both deaths.” Office of the Dis. Atty., Press Release, Jan. 24, 2024, https://perma.cc/4FX8-3K6G. Admission followed an “extensive” pretrial hearing on results obtained with a “MiSeq FGx System.” Id. This “instrument [can] interrogate up to 96 combined SNP and STR libraries in a single run” using a massively parallel sequencing-by-synthesis procedure. Verogen MiSeq FGx Sequencing System for Next-generation Sequencing of Forensic Genomics Libraries, https://perma.cc/LUF7-YMSP.
fragments are shorter than the STR alleles.94 Indeed, even with undegraded samples, STR analysts must cope with phenomena such as stutter peaks that are near a true peak but might be confused with a peak from a different allele. SNP analysis would obviate this problem with electrophoretic-STR data.
On the other hand, a SNP locus has fewer alleles than an STR locus (usually only two). Hence, it takes more of them to achieve high levels of discrimination. MPS systems solve this problem because they can analyze many separate SNP loci at once. Data on how frequently major SNP alleles occur in various population groups have been collected, so the significance of a match in a multilocus profile can be assessed numerically.95 As such, some scientists see STR profiling as outdated—but recommend including STRs in the sequencing process if only to avoid having to redo the DNA databases of STR profiles of convicted offenders and arrestees.
Recent work with SNPs for individual identification focuses on using more than one SNP at a time for a marker. Two or three SNPs can be chosen within a short segment of DNA (smaller than 300 base pairs, for example). Such short blocks of DNA are very likely to be inherited as a package from one parent, and MPS targeted to them can identify this “microhaplotype” in a single sequence run. A pair of SNPs has more possible types than a single SNP, so each microhaplotype is more informative than a single-SNP locus. Panels of microhaplotypes have been studied for individual identification and other forensic-science applications.96 With a sufficient number of SNPs, the power to discriminate between two randomly selected individuals is superior to that of conventional STR profiling.97
94. Moreover, SNPs have advantages for forensic tests that do not make direct comparisons of samples of known (from a suspect, for example) and unknown (from a crime scene) origin. They tend to be specific to certain populations, so they can be used to infer ancestry. See infra section titled “Biogeographic Ancestry Testing.” Most have far lower mutation rates, which is advantageous in parentage and other kinship testing.
95. See infra sections titled “Could a Close Relative Be the Source?” and “Frequencies, Probabilities, and Prejudice.”
96. To reduce the possibility of conveying significant information about disease status or susceptibility, microhaplotype loci can be limited to SNPs located far from genes. Ones that are very close to certain genes would tend to be inherited along with the gene, potentially allowing them to be used as markers for disease-causing gene mutations.
97. E.g., Kenneth K. Kidd et al., Evaluating 130 Microhaplotypes Across a Global Set of 83 Populations, 29 Forensic Sci. Int’l: Genetics 29 (2017), https://doi.org/10.1016/j.fsigen.2017.03.014; Aliye Kureshi et al., Construction and Forensic Application of 20 Highly Polymorphic Microhaplotypes, 7 Royal Soc’y Open Sci. 191937 (2020), https://doi.org/10.1098/rsos.191937; Jing-Bo Pang et al., A 124-plex Microhaplotype Panel Based on Next-generation Sequencing Developed for Forensic Applications, 10 Sci. Reps. 1945 (2020), https://doi.org/10.1038/s41598-020-58980-x; Andrew J. Pakstis et al., The Population Genetics Characteristics of a 90 Locus Panel of Microhaplotypes, 140 Hum. Genetics 1753 (2021), https://doi.org/10.1007/s00439-021-02382-0.
Summary
DNA contains the genetic information of an organism. In humans, most of the DNA is found in the cell nucleus, where it is organized into separate chromosomes. Each chromosome is like a book, and each cell has the same set of books of various sizes. There are two copies of each book (a “diploid” genome)—one copy from the father, one from the mother (the two “haploid” genomes). Thus, there are two copies of the book entitled “Chromosome One,” two copies of “Chromosome Two,” and so on. The text is mostly the same in a pair of books, but sexual reproduction results in important differences as well as inconsequential ones.
Genes are like paragraphs in the books. They encode different proteins and RNAs. Parts of the DNA text appear to have no coherent message, but some of the noncoding DNA affects the production of the gene products. Two individuals sometimes have different versions (alleles) of the same gene as well as variants of the text outside of (or interrupting) the genes. Some alleles result from the substitution of one letter for another. These are SNPs. Others come about from the insertion or deletion of single letters, and still others represent a kind of stuttering repetition of a string of extra letters. These are the VNTRs and STRs.98 The locations within a chromosome where these interpersonal variations occur are called loci.
Historically, forensic DNA typing relied primarily on the length differences of a small number of VNTR and then a larger number of STR loci.99 STR profiling depends on a chemical reaction for generating a great many copies of DNA segments (PCR amplification), followed by separation of the copies according to their length (capillary electrophoresis). We can refer to this well-established technology as PCR-CE for STRs, or even more compactly, as STR-CE profiling. Modern sequencing technology enables more and improved SNP-based loci for identity determinations and other forensic-science applications.
What Establishes That a Genetic System Is Valid for Identification?
Regardless of the kind of genetic system used for typing—STR, SNP, or still other polymorphisms—some general principles and questions can be applied to judge its validity.100 First, the nature of the polymorphism should be well
98. In addition to the 23 pairs of “books” (chromosomes) in the cell nucleus, other scraps of DNA “text” reside in each of the mitochondria, the power plants of the cell. See section titled “Mitochondrial DNA” below.
99. For a short time, a few SNPs in protein-coding DNA also were used.
100. In addition to the suggestions on studying validity in this section, see section titled “Validation” below.
characterized. Is it a sequence polymorphism or a length polymorphism? Where is it situated within the genome? This information should be in the published literature or in archival genome databanks.
Second, the published scientific literature can be consulted to verify claims that a particular method of analysis can produce accurate profiles under various conditions. Ideally, these claims will rest on a detailed understanding of how the analytical procedure works—that is, of how measurements are made and how inferences are drawn from these measurements—as well as empirical studies of the results achieved with the methods under experimental conditions or in casework.101 Although such validation studies have been conducted for all the systems described here, determining the point at which the validation of a particular system is sufficiently extensive and convincing to pass scientific muster may well require expert assistance.102 The standards for validation that have been issued by private standards-developing organizations or expert groups often have only general requirements.103
Finally, the population genetics of the system should be characterized. As new systems are discovered, researchers typically analyze convenient collections of DNA samples from various human populations and publish studies of the relative frequencies of each allele in these population samples.104 These studies give a measure of the extent of variability at the polymorphic locus in the various populations, and thus of the potential probative power of the marker for distinguishing among individuals.
At this point, the capability of the widely used PCR-CE procedures to ascertain STR genotypes accurately cannot be doubted.105 But the technology has its limits, and a laboratory should be able to show that it is operating within the limits
101. See id.
102. See section titled “Relevant Expertise” above.
103. When it comes to specifying what level of performance an analytical method must be shown to possess, many standards are vague or vacuous. For instance, the FBI’s Scientific Working Group on DNA Analysis Methods conflates accuracy with repeatability and reproducibility of measurements and cursorily asserts that these properties “should be evaluated.” SWGDAM, Validation Guidelines for DNA Analysis Methods §§ 3.5.1 & 3.5.2 (Dec. 5, 2016). ANSI/ASB Standard 038 (2020), entitled “Standard for Internal Validation of Forensic DNA Analysis Methods,” likewise contains no minimum levels for accuracy and reproducibility, and its “requirements” barely go beyond the command that “[t]he laboratory shall conduct internal validation studies on all forensic DNA analysis methodologies prior to implementation.” More detailed standards for particular techniques are in progress.
104. On the populations that should be sampled, see section titled “Could an Unrelated Person Be the Source?” below.
105. See, e.g., President’s Council of Advisors on Sci. & Tech. (PCAST), Report to the President: Forensic Science in Criminal Courts: Ensuring Scientific Validity of Feature-Comparison Methods, Sept. 2016, at 75, https://perma.cc/R76Y-7VU (“single-source samples or simple mixtures of two individuals, such as from many rape kits, is an objective method that has been established to be foundationally valid”); accord, John M. Butler et al., DNA Mixture Interpretation: A NIST Scientific Foundation Review, at 3 & 7, June 2021, NISTIR 8351-DRAFT; see also supra Introduction.
for which it has been tested (or if not, to explain why the change in circumstances does not matter). If small samples are at issue, it will be important to ask what validity studies show about the performance of the system as sample size is reduced. If samples include DNA mixtures, the question of whether the validity studies show that the method works well enough with similarly complex samples will arise.106
As next-generation technologies are introduced in court, the same basic questions will need to be answered: What is the principle of the new technology? Is it simply an extension of existing technologies, or does it invoke entirely new concepts? Is the new technology used in research or clinical applications independent of forensic science? If so, are there grounds for concern that the new technology has limitations that might affect its application in the forensic sphere? Finally, what testing has been done to establish that the new technology is valid and reliable when used on forensic samples? For massively parallel sequencing and microarray technologies, the questions may be directed as well to the bioinformatics methods used to analyze and interpret the raw data. Obtaining answers to these questions will likely require input both from experts involved in technology development and application and from knowledgeable forensic experts.
Of course, the fact that scientists have shown that it is possible to extract DNA and to analyze it in a way that bears on the issue of identity does not mean that a particular laboratory has adopted a suitable protocol, is generally proficient in following it, and has implemented the procedure correctly in the case at bar. These more case-specific issues are considered next.
Sample Collection and Laboratory Performance
Sample Collection and Contamination
The primary determinants of whether DNA typing can be done on any particular sample are (1) the quantity of DNA present in the sample and (2) the extent to which it is degraded. Generally speaking, if a sufficient quantity of reasonable-quality DNA can be extracted from a crime-scene sample, no matter what the nature of the sample, DNA typing can be done. Thus, DNA typing has been performed on old blood stains, semen stains, vaginal swabs, hair, bone, bite marks, cigarette butts, drinking cups, guns, garments, urine, and fecal material. This section discusses what constitutes sufficient quantity and reasonable quality in the context of capillary electrophoresis of PCR-amplified short tandem repeats (STR-CE profiling), which has been the predominant method for forensic DNA identification for more than thirty years. Complications from
106. See section titled “Validation of PGS Programs” below.
contaminants and inhibitors also are discussed, and emerging alternatives to STR-CE profiling are noted. The treatment of samples that contain DNA from two or more contributors is discussed in the section titled “Mixtures” below.
Did the Sample Contain Enough DNA?
Amounts of DNA present in some typical kinds of samples vary from a trillionth or so of a gram (a picogram) for a hair shaft to several millionths of a gram (micrograms) for a postcoital vaginal swab.107 Most PCR test protocols recommend samples on the order of 1 billionth of a gram (1 nanogram) for optimal yields. Normally, the number of amplification cycles for nuclear DNA is limited to 28, 29, or 30 to avoid detecting alleles from less than about 10 to 15 cell equivalents of DNA (66 to 100 picograms).108
Procedures for STR-CE typing of still smaller samples—down to a single cell’s worth of nuclear DNA—have been studied. These have been shown to work, to some extent, with trace or contact DNA left on the surface of an object such as the steering wheel of a car.109 Improved reaction chemistries, amplification to a greater number of PCR cycles,110 and detection systems that provide greater sensitivity have made detection of PCR products associated with single
107. A combination of chemical and physical methods permits DNA to be isolated from the other chemicals in a specimen collected from a crime scene or an individual. The basics are fairly standard in academic research (see generally Akash Gautam, DNA and RNA Isolation Techniques for Non-Experts (2022)), but scientists in a forensic DNA laboratory often face a high volume of samples, a variety of sample types, limited quantities of biological material, DNA degradation, chemical inhibitors, and environmental contaminants that can interfere with later analysis. Hence, there is no single best DNA extraction method. The method of choice often depends on the biological sample concerned. Kevin Wai Yin Chong et al., Recent Trends and Developments in Forensic DNA Extraction, 3 WIREs Forensic Sci. e1395 (2020), https://doi.org/10.1002/wfs2.1395. To save time and to avoid the loss of DNA in the extraction, isolation, and quantitation process, direct PCR amplification techniques have been developed. See, e.g., Sarah E. Cavanaugh & Abigail S. Bathrick, Direct PCR Amplification of Forensic Touch and Other Challenging DNA Samples: A Review, 32 Forensic Sci. Int’l: Genetics 40 (2018), https://doi.org/10.1016/j.fsigen.2017.10.005; Belinda Martin et al., Comparison of Six Commercially Available STR Kits for Their Application to Touch DNA Using Direct PCR, 4 Forensic Sci. Int’l: Reports 100243 (2021), https://doi.org/10.1016/j.fsir.2021.100243.
108. But see SWGDAM, Guidelines for STR Enhanced Detection Methods 2 (Oct. 6, 2014), https://perma.cc/NYF7-R9HH (“current . . . kits . . . entail 26 to 32 cycles of PCR amplification with 0.5 to 2 ng of DNA template”).
109. On the cellular material deposited by contact with hands, see Julia Burrill et al., Corneocyte Lysis and Fragmented DNA Considerations for the Cellular Component of Forensic Touch DNA, 51 Forensic Sci. Int’l: Genetics 102428 (2021), https://doi.org/10.1016/j.fsigen.2020.102428.
110. STR-CE testing of LT-DNA samples with “extra” PCR cycles is unusual in the United States. The laboratory of the Office of the Chief Medical Examiner for New York City employed a 31-cycle protocol instead of the then-established 28 cycles that generally withstood objections at the trial court level. In 2020, however, the New York Court of Appeals held in People v. Williams, 147 N.E.3d 1131 (N.Y. 2020), that, in light of defendant’s showing that there was a controversy
molecules of DNA commonplace in casework analysis.111 In addition, laboratory studies have shown that PCR-related methods of whole-genome amplification can make copies of a single DNA molecule that then may yield usable STR profiles with STR-CE.112
But when PCR is used on such small samples of DNA, chance effects might result in one allele being amplified much more than another. Alleles can drop out, small peaks from unusual alleles at other loci can appear, stutter peaks tend to be larger in relation to their parent peak, and bits of extraneous DNA can contribute to the profile. Laboratories normally conduct experiments with samples at successively lower concentrations not only to determine an “analytical threshold” at which a true peak reliably can be distinguished from low-level “noise” but also to establish a higher “stochastic threshold” at which the sporadic effects become apparent.113 Laboratory protocols for acquiring further data and interpreting electropherograms from samples that exhibit stochastic effects vary and are discussed in connection with the interpretation of mixed DNA samples and the coming of age of probabilistic genotyping software (see section titled “Mixtures” below).
Although there are tests to estimate the quantity of DNA in a sample, whether a particular sample contains enough human DNA to allow PCR-based typing cannot always be predicted accurately, and it is not scientifically necessary to apply an arbitrary threshold as a prerequisite for testing.114 If a result is
among scientists on the use of the additional cycles, it was an abuse of discretion to deny his motion for a pretrial hearing on the general acceptance of the modified PCR procedure.
In response to criticism of the LT-DNA evidence in the unsuccessful prosecution of Sean Hoey for a terrorist bombing in Omagh, Northern Ireland, England briefly suspended its use of the Forensic Science Service’s 34-cycle procedure. Duncan Campbell & Vikram Dodd, Police Suspend Use of Discredited DNA Test After Omagh Acquittal, Guardian, Dec. 22, 2007. After it reinstated the method (with an added quantification step), the English Court of Appeal opined that “Low Template DNA can be used to obtain profiles capable of reliable interpretation if the quantity of DNA that can be analysed is above the stochastic threshold [of] between 100 and 200 picograms.” R. v. Reed, [2009] (CA Crim. Div.) EWCA Crim. 2698, ¶ 74; Denise Syndercombe Court, Low Copy Number DNA: Where Next?, 50 Med., Sci. & Law 55 (2010), https://doi.org/10.1258/msl.2010.010015.
111. David Moore et al., A Comprehensive Study of Allele Drop-In over an Extended Period of Time, 48 Forensic Sci. Int’l: Genetics 102332 (2020), https://doi.org/10.1016/j.fsigen.2020.102332.
112. Man Chen et al., Comparison of CE- and MPS-based Analyses of Forensic Markers in a Single Cell After Whole Genome Amplification, 45 Forensic Sci. Int’l: Genetics 102211 (2020) https://doi.org/10.1016/j.fsigen.2019.102211; Richard Jäger, New Perspectives for Whole Genome Amplification in Forensic STR Analysis, 23 Int’l J. Molecular Sci. 7090 (2022), https://doi.org/10.3390/ijms23137090; Xu Qiannan et al., Evaluating the Effects of Whole Genome Amplification Strategies for Amplifying Trace DNA Using Capillary Electrophoresis and Massive Parallel Sequencing, 56 Forensic Sci. Int’l: Genetics 102599 (2022) https://doi.org/10.1016/j.fsigen.2021.102599.
113. See John M. Butler, Advanced Topics in Forensic DNA Typing: Interpretation 40–44 & 93–95 (2015).
114. DNA concentration levels such as 100 or 200 picograms have been suggested to differentiate a conventional DNA profile from a low-level target profile. See supra note 110. However, STR-CE technology has improved since these numbers were proposed (see Davis R.L. Watkins
obtained, and if the controls (samples of known DNA and blank samples) have behaved properly, then the sample had enough DNA.115 This is so whether or not the label low-copy number (LCN) or low template (LT) applies.116
Was the Sample of Sufficient Quality?
The primary determinant of DNA quality for forensic analysis is the extent to which the long DNA molecules are intact. Within the cell nucleus, each molecule of DNA extends for millions of base pairs. Outside the cell, DNA spontaneously degrades into smaller fragments at a rate that depends on temperature, exposure to oxygen, and, most importantly, the presence of water.117 In dry biological samples,
et al., Revisiting Single Cell Analysis in Forensic Science, 11 Sci. Reps. 7054 (2021), https://doi.org/10.1038/s41598-021-86271-6, & authorities cited), and “[t]hresholds are often difficult to apply in a meaningful way, for example, in a sample that comprises DNA from two or more individuals, the total quantifiable does not reflect the individual contributions.” Forensic Science Regulator (U.K.), Guidance: The Interpretation of DNA Evidence (Including Low-Template DNA), FSR-G-202 § 5.2.4 (2020), https://perma.cc/TX2Y-5SXL.
115. Different views have been expressed on the desirability of performing replicate tests (splitting the sample into several PCR tubes) and on how to interpret the resulting profiles. The “consensus method” discards all possible alleles other than those that are detected in two or more of the resulting electropherograms. Simon Cowen et al., An Investigation of the Robustness of the Consensus Method of Interpreting Low-Template DNA Profiles, 5 Forensic Sci. Int’l: Genetics 400 (2011), https://doi.org/10.1016/j.fsigen.2010.08.010. As statistical methods for modeling the sources of variability have advanced, replicate testing has fallen out of favor. See, e.g., David J. Balding, Evaluation of Mixed-source, Low-template DNA Profiles in Forensic Science, 110 Proc. Nat’l Acad. Sci. 12241 (2013), https://doi.org/10.1073/pnas.1219739110; Forensic Science Regulator, supra note 114, § 11.1.4. Replicate profiling of small samples continues to be discussed. E.g., Forensic Science Regulator, supra, § 12; Simone Gittelson et al., Low-template DNA: A Single DNA Analysis or Two Replicates?, 264 Forensic Sci. Int’l 139 (2016), https://doi.org/10.1016/j.forsciint.2016.04.012; SWGDAM, supra note 108, § 4.1.
116. The phrase “low copy number” originally was coined for the Forensic Science Service’s method that used 34 PCR cycles. It has caused confusion. E.g., United States v. McCluskey, 954 F. Supp. 2d 1224, 1278 (D.N.M. 2013) (skirting the debate between the parties on whether LCN refers to modifications in the testing or to the quantity of DNA by focusing on “whether the Government’s DNA results in this case are reliable and admissible” rather than “establish[ing] a definition of ‘LCN testing’”); United States v. Davis. 602 F. Supp. 2d 658 (D. Md. 2009) (avoiding “making a finding with regard to the dueling definitions of LCN testing advocated by the parties”); State v. Bigger, 254 P.3d 1142, 1151–52 (Ariz. Ct. App. 2011) (“We need not determine the proper definition for low copy number, or whether the DNA testing used in this case is labeled properly as LCN.”). The more generic term “low template” (LT) DNA analysis includes the use of longer injection times for CE and sample concentration methods. Forensic Science Regulator, supra note 114, § 5.2.1. SWGDAM, supra note 108, introduced a more complicated definition.
117. Other forms of chemical alteration to DNA are well studied, both for their intrinsic interest and because chemical changes in DNA are a contributing factor in the development of cancers in living cells. Some forms of DNA modification, such as those produced by exposure to ultraviolet radiation, inhibit the amplification step in PCR-based tests, whereas other chemical
protected from air and not exposed to temperature extremes, DNA degrades very slowly. STR testing has proved effective with old and badly degraded material such as the remains of the Tsar Nicholas family (buried in 1918 and recovered in 1991).118
The extent to which degradation affects a PCR-based test depends on the size of the DNA segment to be amplified. For example, in a sample in which the bulk of the DNA has been degraded to fragments well under 1,000 base pairs in length, it may be possible to amplify a 100-base-pair sequence, but not a 1,000-base-pair target. Consequently, the shorter alleles may be detected in a highly degraded sample, but the larger ones may be missed. Inasmuch as the size differences among STR alleles at a locus are quite small (typically no more than 50 base pairs), “locus dropout” is more common than the dropout of only one allele at a heterozygous locus.119
DNA can be exposed to a great variety of environmental insults without any effect on its capacity to be typed correctly. Exposure studies have shown that contact with a variety of surfaces, both clean and dirty, and with gasoline, motor oil, acids, and alkalis either have no effect on DNA typing or, at worst, render the DNA untypable.120
Although contamination with microbes generally does little more than degrade the human DNA, other problems sometimes can occur. Therefore, the validation of DNA typing systems should include tests for interference with a variety of microbes to see if artifacts occur. If artifacts are observed, then control tests should be applied to distinguish between the artifactual and the true results.
modifications appear to have no effect. Cydne L. Holt et al., TWGDAM Validation of AmpFlSTR PCR Amplification Kits for Forensic DNA Casework, 47 J. Forensic Sci. 66 (2002), https://doi.org/10.1520/jfs15206j; George F. Sensabaugh & Cecilia von Beroldingen, The Polymerase Chain Reaction: Application to the Analysis of Biological Evidence, in Forensic DNA Technology 63 (Mark A. Farley & James J. Harrington eds., 1991).
118. Peter Gill et al., Identification of the Remains of the Romanov Family by DNA Analysis, 6 Nature Genetics 130 (1994), https://doi.org/10.1038/ng0294-130.
119. In particular, in cases involving severe degradation, loci yielding products greater than 200 base pairs may not be detected. Holt et al., supra note 117. Special primers and very short STRs give better results with extremely degraded samples. See Michael D. Coble & John M. Butler, Characterization of New MiniSTR Loci to Aid Analysis of Degraded DNA, 50 J. Forensic Sci. 43 (2005), https://doi.org/10.1520/jfs2004216. Sequencing methods can be even more effective for such samples. See supra section titled “SNPs as identification loci.”
120. Holt et al., supra note 117. Most of the effects of environmental insult readily can be accounted for in terms of basic DNA chemistry. For example, some agents produce degradation or damaging chemical modifications. Other environmental contaminants inhibit restriction enzymes or PCR. (This effect sometimes can be reversed by cleaning the DNA extract to remove the inhibitor.) But environmental insult does not result in the selective loss of an allele at a locus or in the creation of a new allele at that locus.
Laboratory Performance
What Quality Control and Assurance Measures Are in Place?
DNA profiling of suitable samples is valid and reliable when properly conducted, but confidence in a particular result also depends on the quality control and quality assurance procedures in the laboratory. Quality control refers to measures to help ensure that DNA-typing results and their interpretation meet a specified standard of quality. Quality assurance refers to monitoring, verifying, and documenting laboratory performance. A quality assurance program helps demonstrate that a laboratory is meeting its quality control objectives.121
Professional bodies within forensic science have described general procedures for quality assurance. Guidelines for DNA analysis have been prepared by FBI-appointed groups (the current incarnation is known as the Scientific Working Group on DNA Analysis Methods [SWGDAM]),122 the federally supported Organization of Scientific Area Committees for Forensic Science (OSAC),123 and private Standards Developing Organizations (SDOs).124
121. For a review of the history of quality assurance in forensic DNA testing, see Joseph L. Peterson et al., The Feasibility of External Blind DNA Proficiency Testing. I. Background and Findings, 48 J. Forensic Sci. 21, 22 (2003), https://doi.org/10.4324/9780203380826.
122. The FBI established the Technical Working Group on DNA Analysis Methods (TWGDAM) in 1988 to develop standards. The DNA Identification Act of 1994, 34 U.S.C. §§ 12591(a)–(c), 14131(a) & (c), created a DNA Advisory Board (DAB) to assist in promulgating quality assurance standards, but the legislation allowed the DAB to expire after five years (unless extended by the director of the FBI). 34 U.S.C. § 12591(b)(4). TWGDAM functioned under the DAB, 34 U.S.C. § 12591(a)(4), and was renamed the Scientific Working Group on DNA Analysis Methods (SWGDAM) in 1999. When the FBI allowed DAB to expire, SWGDAM replaced it. See Norah Rudin & Keith Inman, An Introduction to Forensic DNA Analysis 180 (2d ed. 2002); Paul C. Giannelli, Regulating Crime Laboratories: The Impact of DNA Evidence, 15 J.L. & Pol’y 59, 82–83 (2007). SWGDAM characterizes the FBI quality-assurance standards as establishing minimum requirements that it supplements with more detailed, noncompulsory guidelines. SWGDAM, Frequently Asked Questions, https://perma.cc/Z9AR-FG6A, last visited Sept. 9, 2025.
123. The Department of Commerce’s National Institute of Standards and Technology (NIST) supports OSAC’s standards-writing efforts and maintains “a repository of selected published and proposed standards for forensic science . . . to promote valid, reliable and reproducible forensic results.” NIST, OSAC Registry (updated Dec. 22, 2023), https://perma.cc/M9LS-96UN. The OSAC Registry maintains that “the standards on this Registry have undergone a technical and quality review process that actively encourages feedback from forensic science practitioners, research scientists, human factors experts, statisticians, legal experts, and the public.” Id. The standards are voluntary, and some have been criticized as vague or trivial. Geoffrey Stewart Morrison et al., Vacuous Standards—Subversion of the OSAC Standards-development Process, 2 Forensic Sci. Int’l: Synergy 206 (2020), https://doi.org/10.1016/j.fsisyn.2020.06.005. The rigor of the actual review process, particularly as it involves independent scientific and statistical review, also has been questioned. See Kaye et al., supra note 5, § 12.5.1, at 680–82; Maneka Sinha, Radically Reimagining Forensic Evidence, 73 Ala. L. Rev. 879 (2022).
124. The major SDO that publishes voluntary standards for forensic DNA analysis is the Academy Standards Board (ASB) of the American Academy of Forensic Sciences. AAFS, Academy
A small number of states require forensic DNA laboratories to be accredited,125 and federal law requires accreditation or other safeguards for laboratories that receive certain federal funds126 or participate in the national DNA database system.127
Documentation
Quality assurance guidelines normally call for laboratories to document laboratory organization and management, personnel qualifications and training, facilities, evidence control procedures, validation of methods and procedures, analytical procedures, equipment calibration and maintenance, standards for case documentation and report writing, procedures for reviewing case files and testimony, proficiency testing, corrective actions, audits, safety programs, and review of subcontractors.
Of course, maintaining documentation and records alone does not guarantee the correctness of results obtained in any particular case. Measurement error can be estimated but not eliminated, and process errors in analysis or
Standards Board (2022), https://perma.cc/Y8N4-A4FU. Inasmuch as the dominant voice in both OSAC and the SDOs that promulgate forensic-science standards is the practicing forensic-science community, a consensus standard does not necessarily reflect a consensus of experts in the broader scientific community. Within OSAC and the ASB, agreement of two-thirds or more of the eligible voters constitutes a consensus. The votes are not published.
125. Joseph Peterson & Matthew Hickman, Forensic Science Practice in the United States, in The Global Practice of Forensic Science 301, 331 (Douglas H. Ubelaker ed., 2015). New York was the first state to impose this requirement. N.Y. Exec. Law § 995-b (McKinney 2006) (requiring accreditation by the state Forensic Science Commission). Only one state, Texas, requires its forensic analysts to be licensed. Texas Forensic Sci. Comm’n, Forensic Analyst Licensing Program, https://perma.cc/6UDT-NU3W, last visited Sept. 9, 2025.
126. The Justice for All Act, enacted in 2004, required DNA labs to be accredited within two years “by a nonprofit professional association of persons actively involved in forensic science that is nationally recognized within the forensic science community” and to “undergo external audits, not less than once every 2 years, that demonstrate compliance with standards established by the Director of the Federal Bureau of Investigation.” 34 U.S.C. § 12592(b)(2)(A). The 2004 Act also requires applicants for federal funds for forensic laboratories to certify that the laboratories use “generally accepted laboratory practices and procedures, established by accrediting organizations or appropriate certifying bodies,” 34 U.S.C. § 10562(2), and that “a government entity exists and an appropriate process is in place to conduct independent external investigations into allegations of serious negligence or misconduct substantially affecting the integrity of the forensic results committed by employees or contractors of any forensic laboratory system, medical examiner’s office, coroner’s office, law enforcement storage facility, or medical facility in the State that will receive a portion of the grant amount.” Id. § 10562(4).
127. See 34 U.S.C. § 12592(b)(2) (requiring that records in the database come from laboratories that “have been accredited by a nonprofit professional association . . . and . . . undergo external audits, not less than once every 2 years [and] that demonstrate compliance with standards established by the Director of the Federal Bureau of Investigation”).
interpretation can occur as a result of a deviation from an established procedure, an analyst misjudgment, or an accident. Although administrative and technical review procedures within a laboratory should be designed to detect errors before a report is issued, it is always possible that some incorrect result will slip through.128 Accordingly, determination that a laboratory maintains a strong quality-assurance program, either on paper or in practice, does not guarantee accuracy in all cases.129
Validation
The validation of procedures is central to quality assurance. Developmental validation is undertaken to determine the applicability of a new test or equipment to crime-scene samples; it defines conditions that give accurate results and identifies the limitations of the procedure.130 For example, a new genetic locus being considered for use in forensic analysis will be tested in both fresh samples and in samples typical of those found at crime scenes. The validation would include samples originating from different tissues, samples containing degraded DNA, samples contaminated with microbes, samples containing DNA mixtures, and so on. A valid typing method usually will report a genotype that is in a test sample and usually will not report a genotype that is not in the test sample.131 Developmental validation of a new set of loci also includes the generation of population databases and the testing of alleles for statistical independence. Developmental validation normally results in publication in the scientific literature and reflects scientific norms for theoretical and
128. See U.S. Dep’t of Just., Office of the Inspector Gen., The FBI DNA Laboratory: A Review of Protocol and Practice Vulnerability (2004), available at https://oig.justice.gov/reports/fbi-dna-laboratory-review-protocol-and-practice-vulnerabilities.
129. The District of Columbia’s independent, state-of-the-art Department of Forensic Sciences twice lost its accreditation—and each of its first two directors—following complaints from prosecutors, first about DNA mixture analyses and later about firearms-toolmark examinations. See Keith L. Alexander & Julie Zauzmer, Director of D.C.’s Embattled DNA Lab Resigns After Suspension of Testing, Wash. Post, May 1, 2015, at B1, https://perma.cc/Z4FM-EWWB; Editorial, The D.C. Crime Lab Is in Trouble—Again, Wash. Post, June 4, 2021, at A20.
130. See supra section titled “What Establishes That a Genetic System Is Valid for Identification?”
131. No test is always correct. For a set of test samples, all of which contain a particular genotype, a test might be positive for that genotype in, say, 96% of them. This proportion is the observed “sensitivity” in the test set. For a set of test samples, none of which contain the genotype, the test might be negative for that genotype in, say, 99% of them. This proportion is the observed “specificity” for that genotype in the test set. A perfect test would have 100% sensitivity and 100% specificity in all properly conducted experiments. For further discussion of the validity of binary (present-or-absent) testing, see Kaye & Stern, supra note 12.
empirical support of claims for methods,132 but the empirical aspects for validating a new procedure can be accomplished in multiple laboratories well ahead of publication.133
So-called internal validation, on the other hand, involves the capacity of a specific laboratory to analyze the new loci or use the new methodology or equipment.134 The laboratory should verify that it can reliably perform an established procedure that already has undergone developmental validation.135 In particular, before adopting a new procedure, the laboratory should verify that its analysts can obtain correct results with samples that are representative of casework.
Proficiency testing
Proficiency testing in forensic genetic testing is designed to ascertain whether an analyst can correctly determine genetic types in a sample whose origin is unknown to the analyst but is known to a tester. Proficiency is demonstrated by making correct determinations in repeated trials. The laboratory also can be tested to verify that it correctly computes random-match probabilities or likelihood ratios.
An internal proficiency trial is devised and conducted within a laboratory.136 One person in the laboratory prepares the sample and then administers the test to another person in the laboratory. In an external trial, the test sample originates
132. As such, validation is not easily reduced to standards defining the research that needs to be undertaken, and validation standards often seem open textured. See supra note 103.
133. SWGDAM “encourage[s]” but does not require publication of developmental validity studies in scientific venues. SWGDAM, supra note 103, § 2.2.1.2. Whether the validity required for admission of scientific evidence can be established in the absence of a robust set of scientific publications is a legal rather than a strictly scientific question. However, it has been argued that scientific norms demand multiple studies with confirmation coming from separate research groups rather than only from the developer of a method. President’s Council of Advisors on Sci. & Tech., supra note 105, at 80.
134. This line between developmental and internal validation is not always clear. Successful internal validation efforts add to the body of knowledge demonstrating that an analytical method performs as it should and thus enhances or extends what might be seen as a weak but encouraging previous validation. Furthermore, “internal validation” sometimes designates finding information needed to fine-tune a method. That function is better referred to as calibration.
135. Validation builds on the accumulated body of knowledge and experience. Thus, some aspects of validation testing need be repeated only to the extent required to verify that previously established principles apply.
136. Some forensic-science organizations do not use the term “proficiency testing” for tests of examiner performance developed or conducted within a single laboratory. In their view, proficiency testing requires interlaboratory comparisons. OSAC, OSAC Preferred Terms 2 (Aug. 2022), https://perma.cc/KJC4-6HWE.
from outside the laboratory—from another laboratory, a commercial vendor, or a regulatory agency. In a declared (or open) proficiency trial, the analyst knows the sample is a proficiency sample. The DNA Identification Act of 1994 required proficiency testing for analysts in the FBI as well as those in laboratories participating in the national database or receiving federal funding,137 but these matters are now left to the discretion of the director of the FBI.138 The standards of accrediting bodies typically call for periodic open, external proficiency testing.139
In a blind or, more properly, “full blind” trial, the sample is submitted so that the analyst does not recognize it as a proficiency sample. A full-blind trial provides a better indication of proficiency because it ensures that the analyst will not give the trial sample any special attention, and it tests more steps in the laboratory’s processing of samples. However, full-blind proficiency trials entail considerably more organizational effort and expense than open proficiency trials. Obviously, the “evidence” samples prepared for the trial have to be sufficiently realistic that the laboratory does not suspect the legitimacy of the submission. A police agency and prosecutor’s office have to submit the “evidence” and respond to laboratory inquiries with information about the “case.” Finally, the genetic profile from a proficiency test must not be entered into regional and national databases. Consequently, although some forensic DNA laboratories participate in full-blind testing, they are not required to do so.140
137. Pub. L. 103–22 § 210304(b)(2) (codified as amended 34 U.S.C. § 12592(b)(2)(A)(ii)) (requiring external proficiency testing of laboratories for participation in the national database); id. § 210305(a)(1)(A) (same for FBI examiners).
138. 34 U.S.C. § 12592(b)(2)(A)(ii) (requiring “external audits, not less than once every 2 years, that demonstrate compliance with standards established by the Director of the Federal Bureau of Investigation”). The standards require periodic external proficiency testing. FBI, Quality Assurance Standards for Forensic DNA Testing Laboratories § 13.1 (2020), https://le.fbi.gov/file-repository/forensic-qas-070120.pdf.
139. See Peterson et al., supra note 121, at 24 (describing the ASCL-LAB standards). Certification by the American Board of Criminalistics as a specialist in forensic-biology DNA analysis requires one proficiency trial per year. Accredited laboratories must maintain records documenting compliance with required proficiency-test standards.
140. However, laboratory management can blind a particular analyst within the laboratory by injecting disguised “cases” into the usual flow of cases. Section 210303(c) of the DNA Identification Act of 1994, Pub. L. 103–22, 108 Stat. 2069, required the director of the National Institute of Justice to report to Congress on the feasibility of establishing an external blind-proficiency-testing program for DNA laboratories. A National Forensic DNA Review Panel advised the director that “blind proficiency testing is possible, but fraught with problems” of the kind listed above. Peterson et al., supra note 121, at 30. It “recommended that a blind proficiency testing program be deferred for now until it is more clear how well implementation of the first two recommendations [the promulgation of guidelines for accreditation, quality assurance, and external audits of casework] are serving the same purposes as blind proficiency testing.” Id.
How Are Samples Created and Handled?
Sample mishandling, mislabeling, or contamination, whether in the field or in the laboratory, is more likely to compromise a DNA analysis than is an error in genetic typing. For example, a sample mix-up due to mislabeling reference blood samples taken at the hospital could lead to an incorrect association of crime-scene samples to a reference individual or to incorrect exclusions. Similarly, packaging two items with wet blood stains into the same bag could result in a transfer of stains between the items, rendering it difficult or impossible to determine whose blood was originally on each item. Contamination in the laboratory may result in artifactual typing results or in the incorrect attribution of a DNA profile to an individual or to an item of evidence. Procedures should be prescribed and implemented to guard against such error.
Mislabeling or mishandling can occur when biological material is collected in the field, when it is transferred to the laboratory, when it is in the analysis stream in the laboratory, when the analytical results are recorded, or when the recorded results are transcribed into a report. Mislabeling and mishandling can happen with any kind of physical evidence and are of great concern in all fields of forensic science. Checkpoints should be established to detect mislabeling and mishandling along the line of evidence flow. Investigative agencies should have guidelines for evidence collection and labeling so that a chain of custody is maintained. Similarly, there should be guidelines, produced with input from the laboratory, for handling biological evidence in the field.
Professional guidelines and recommendations require documented procedures to ensure sample integrity and to avoid sample mix-ups, labeling errors, recording errors, and the like. They also mandate case review to identify inadvertent errors before a final report is released. Finally, laboratories must retain, when feasible, portions of the crime-scene samples and extracts to allow reanalysis.141 However, retention is not always possible. For example, retention of original items is not to be expected when the items are large or immobile (for example, a wall or sidewalk). In such situations, a swabbing, scraping, or cutting of the stain from the item would typically be collected and retained. There also are situations where the sample is so small that it will be consumed in the analysis.
141. See FBI, supra note 138, § 7.4.1. Furthermore, failure to preserve potentially exculpatory evidence has been treated as a denial of due process and grounds for suppression. People v. Nation, 604 P.2d 1051, 1054–55 (Cal. 1980). In Arizona v. Youngblood, 488 U.S. 51 (1988), however, the Supreme Court held that a police agency’s failure to preserve evidence not known to be exculpatory does not constitute a denial of due process unless “bad faith” can be shown. Ironically, DNA testing that was not available at Youngblood’s trial established that he had been falsely convicted. Maurice Possley, DNA Exonerates Inmate Who Lost Key Test Case: Prosecutors Ruined Evidence in Original Trial, Chi. Trib., Aug. 10, 2000, at 6, https://perma.cc/F78E-E8K7.
Assuming that appropriate chain-of-custody and evidence-handling protocols are in place, the critical question is whether there are deviations in a particular case. Answering this question may require a review of the total case documentation as well as the laboratory findings. In addition, when retesting original evidence items or the material extracted from them is possible, it can be used to guard against error from mislabeling and mishandling. Should mislabeling or mishandling have occurred, reanalysis of the original sample and the intermediate extracts may detect not only the fact of the error but also the point at which it occurred.142
Contamination describes any situation in which foreign material is mixed with a sample of DNA.143 As noted in the section titled “Was the Sample of Sufficient Quality?” above, contamination by nonbiological materials, such as gasoline or grit, can cause test failures, but it is not a source of genetic typing errors. Similarly, contamination with nonhuman biological materials, such as bacteria, fungi, or plant materials, is generally not a problem. These contaminants may accelerate DNA degradation, but they do not generate spurious human genetic types.
The contamination of greatest concern results from the addition of human DNA. This sort of contamination can occur in three ways. First, the crime-scene samples, by their nature, may contain a mixture of fluids or tissues from different individuals. Examples include vaginal swabs collected as sexual-assault evidence and bloodstain evidence from scenes where several individuals shed blood. The analysis of such mixtures is the subject of the “Mixtures” section below.
Second, the crime-scene samples may be inadvertently contaminated in the course of sample handling in the field or in the laboratory. “Elimination databases” of DNA profiles from laboratory personnel as well as police officers and emergency responders who are present at crime scenes can be used to see
142. Of course, retesting cannot correct all errors that result from mishandling of samples, but it is possible in some cases to detect mislabeling at the point of sample collection if the genetic-typing results on a particular sample are inconsistent with an otherwise consistent reconstruction of events. For example, a mislabeling of husband and wife samples in a paternity case might result in an apparent maternal exclusion, a very unlikely event. The possibility of mislabeling could be confirmed by testing the samples for gender and ultimately verified by taking new samples from each party under better-controlled conditions.
143. Cf. Forensic Science Regulator (U.K.), Forensic Science Regulator Guidance: Contamination Controls—Scene of Crime § 1.1.1, at 4 (2023) (FSR-GUI-0016), https://perma.cc/Q4D2-MGJU (“For the purposes of this guidance, contamination is defined as ‘the undesirable introduction of DNA, or biological material containing DNA, to an item/exhibit recovered from an incident scene which is to be recovered/analysed and before a controlled forensic process is started’. This is distinct from the adventitious transfer of biological material to an exhibit that can also occur, usually prior to the exhibit or sample being recovered and before investigative agencies have intervened, this is often referred to as ‘background DNA’.”); Forensic Science Regulator (U.K.), Forensic Science Regulator Guidance: DNA Contamination Controls—Laboratory § 1.1.1, at 5 (2023), FSR-GUI-0018, https://perma.cc/GU96-M769 (same).
if DNA from these individuals might be in the recovered samples.144 Inadvertent contamination of crime-scene DNA with DNA from a reference sample could lead to a false inclusion. Likewise, with low-template DNA samples, secondary transfer—in which some DNA molecules from one person are present for a time on another person and then are deposited in the crime-scene sample—could be falsely incriminating.145 Research into mechanisms of DNA transfer and DNA- and RNA-based testing that might help clarify the origin of the trace DNA is discussed in the section titled “DNA and RNA Typing of Bodily Fluids or Tissues” below.
Third, carryover contamination in PCR-based typing can occur if the amplification products of one typing reaction are carried over into the reaction mix for a subsequent PCR reaction. If the carryover products are present in sufficient quantity, they could be preferentially amplified over the target DNA. To protect against carryover contamination, the primary strategy is to keep PCR products away from sample materials and test reagents by having separate work areas for pre-PCR and post-PCR sample handling, by preparing samples in controlled-airflow biological safety hoods, by using dedicated equipment (such as pipettes) for each of the various stages of sample analysis, by decontaminating work areas after use (usually by wiping down or by irradiating with ultraviolet light), and by having a one-way flow of sample from the pre-PCR
144. Martine Lapointe et al., Leading-edge Forensic DNA Analyses and the Necessity of Including Crime Scene Investigators, Police Officers and Technicians in a DNA Elimination Database, 19 Forensic Sci. Int’l: Genetics 50 (2015), https://doi.org/10.1016/j.fsigen.2015.06.002; OSAC, Best Practice Recommendations for the Management and Use of Quality Assurance DNA Elimination Databases in Forensic DNA Analysis (Apr. 6, 2021) (2020-N-0007). However, the Genetic Information and Nondiscrimination Act of 2008, 42 U.S.C. § 2000ff et seq., prohibits employers from requesting “genetic information” from employees. An exception, in § 202(b)(6) of the act, applies “where the employer conducts DNA analysis for law enforcement purposes as a forensic laboratory or for purposes of human remains identification, and requests or requires genetic information of such employer’s employees . . . for quality control to detect sample contamination.” 42 U.S.C. § 2000ff-1(b)(6). Non-laboratory police employees have claimed that because they fall outside this exception, the statute protects them from being required to provide elimination samples. In response, it can be argued that the statute’s expansive definition of “genetic test” should be read in light of its main purpose as an anti-discrimination law concerned with health-related genetic tests. David H. Kaye, GINA’s Genotypes, 108 Mich. L. Rev. First Impressions 51 (2010). This intent- or purpose-based approach to the statute was rejected in Lowe v. Atlas Logistics Group Retail Services (Atlanta), 102 F. Supp. 3d 1360 (N.D. Ga. 2015) (construing GINA on the basis of its plain text as prohibiting an employer’s request for a DNA sample for STR profiling to find “a mystery employee [who was] habitually defecating in one of its warehouses”); see also David H. Kaye, EEOC Stays Mum on GINA, Forensic Sci., Stat. & L. (Jan. 22, 2013), https://perma.cc/QS9Z-XAYC.
145. See Peter Gill, Misleading DNA Evidence: A Guide for Scientists, Judges, and Lawyers (2014).
to post-PCR work areas.146 Additional protocols are used to detect carryover contamination.147
In the end, whether a laboratory has conducted proper tests depends both on the general standard of practice and on the questions posed in the particular case. There is no universal checklist, but the selection of tests and the adherence to the correct test procedures can be reviewed by experts and by reference to professional standards.
Inference, Statistics, and Population Genetics in Human Nuclear DNA Testing
What Constitutes a Match? An Exclusion?
The results of DNA testing can be presented in various ways. With discrete allele systems, such as STRs, it is natural—but not essential—to speak of “matching” and “nonmatching” profiles. If the profile obtained from the single-source biological sample taken from the crime scene or the victim (the trace-evidence sample) clearly differs from the DNA in a sample known to come from a particular individual (the reference sample), that individual is excluded as a possible source. At the other extreme, two multilocus genotypes can be clearly identical. In these cases, the DNA evidence typically is quite incriminating,148 but even when two samples have the same genotype, there is a chance that the trace-evidence sample came not from the defendant, but from another individual who has the same genotype. As indicated in the section above titled “A Brief History
146. SWGDAM, Contamination Prevention and Detection Guidelines for Forensic DNA Laboratories (Jan. 12, 2017), https://perma.cc/R2SD-BJ8A.
147. Standard protocols include the amplification of blank control samples—those to which no DNA has been added. If carryover contaminants have found their way into the reagents or sample tubes, these will be detected as amplification products. Outbreaks of carryover contamination can also be recognized by monitoring test results. Detection of an unexpected and persistent genetic profile in different samples indicates a contamination problem. When contamination outbreaks are detected, corrective actions should be taken, and both the outbreak and the corrective action should be documented. Human Forensic Biology Subcomm., Org. of Sci. Area Committees for Forensic Sci., Standard for Interpreting, Comparing and Reporting DNA Test Results Associated with Failed Controls and Contamination Events (OSAC 2020-S-0004, Ver. 2.0, May 2021).
148. Whether being the source of the forensic sample is incriminating and whether someone else being the source is exculpatory depends on the circumstances. For example, a suspect who might have committed the offense without leaving the trace-evidence sample still could be guilty. In a rape case with several rapists, a semen stain could fail to incriminate one assailant because insufficient semen from that individual is present in the sample.
of DNA Evidence,” this complication has produced extensive arguments over the statistical procedures for assessing this possibility.
Some cases lie between the poles of a clear inclusion or exclusion. Since the earliest days of RFLP-VNTR testing, concerns have been expressed about subjective aspects of procedures that leave room for “observer effects”149 in interpreting data.150 When the trace-evidence sample is small and extremely degraded, STR profiling can be afflicted with allelic drop-in, drop-out, and other complications.151 Analysts operating within the framework of reporting that alleles are categorically present or absent then must make judgments as to whether true peaks are missing and whether spurious peaks are present.152 Experts then might disagree about whether a suspect is included or excluded—or whether any conclusion can be drawn.153
What Hypotheses Can Be Formulated About the Source?
In DNA identification cases, the laboratory analyst reports that the sample of DNA from the crime scene and a sample from the defendant have the same genotype. There could be an innocent explanation for the defendant’s DNA being at the crime scene,154 but the matching DNA might not even be the defendant’s. One
149. See generally D. Michael Risinger et al., The Daubert/Kumho Implications of Observer Effects in Forensic Science: Hidden Problems of Expectation and Suggestion, 90 Calif. L. Rev. 1 (2002).
150. E.g., William C. Thompson & Simon Ford, The Meaning of a Match: Sources of Ambiguity in the Interpretation of DNA Prints, in Forensic DNA Technology (Mark J. Farley & James J. Harrington eds., 1990).
151. See supra section titled “Sample Collection and Contamination.”
152. Commentators have proposed that the analyst determine the profile of a trace-evidence sample before knowing the profile of any suspects. E.g., Itiel E. Dror & Jeff Kukucka, Linear Sequential Unmasking-Expanded (LSU-E): A General Approach for Improving Decision Making As Well As Minimizing Noise and Bias, 3 Forensic Sci. Int’l: Synergy 100161 (2021), https://doi.org/10.1016/j.fsisyn.2021.100161; Dan E. Krane et al., Sequential Unmasking: A Means of Minimizing Observer Effects in Forensic DNA Interpretation, 53 J. Forensic Sci. 1006 (2008), https://doi.org/10.1111/j.1556-4029.2008.00787.x.
153. See, e.g., State v. Murray, 174 P.3d 407, 417–18 (Kan. 2008) (inconclusive results were presented as consistent with the defendant’s blood).
154. For example, the two samples would match if someone framed the defendant by putting a sample of defendant’s DNA at the crime scene or in the container of DNA thought to have come from the crime scene. Or, the defendant’s DNA might have made its way to the crime-scene sample inadvertently, as is thought to have happened when a homeless man’s DNA was found on the fingernails of a multi-millionaire found dead in his home after a robbery. After a public defender established that the defendant was in a hospital when the murder occurred, police and prosecutors concluded that his DNA probably had been transferred by paramedics who treated the defendant three hours before they applied a pulse oximeter to the victim’s finger. Katie Worth, Framed for Murder by His Own DNA, Frontline, Apr. 19, 2018, https://perma.cc/C5ST-QWNS.
possibility is laboratory error—the genotypes are not actually the same even though the expert thinks that they are. This situation could arise from mistakes in labeling or handling samples or from cross-contamination of the samples. Another alternative is that the genotypes are truly identical but the forensic sample came from another individual. In general, the true source might be a close relative of the defendant155 or an unrelated person who just happens to have the same profile as the defendant. The former hypothesis we shall refer to as kinship, and the latter as coincidence. To infer that the defendant is the source of the trace-evidence DNA, one must reject these alternative hypotheses of laboratory error, kinship, and coincidence. Table 1 summarizes the logical possibilities.156
Table 1. Hypotheses that Might Explain a Match Between Defendant’s DNA and DNA at a Crime Scene
| IDENTITY | |
| Defendant is source | Same genotype, defendant’s DNA at crime scene |
| NONIDENTITY | |
| Laboratory error | Different genotypes mistakenly found to be the same |
| Kinship | Same genotype, relative’s DNA at crime scene |
| Coincidence | Same genotype, unrelated individual’s DNA at crime scene |
If laboratory error, kinship, and coincidence are rejected as implausible, then only the hypothesis of identity remains. We turn, then, to the considerations that affect the chances of a match when the defendant is not the source of the trace evidence.
Can the Match Be Attributed to Laboratory Error?
Traditional legal and scientific procedures can help to assess the possibilities of errors in handling or analyzing the samples. Scrutinizing the chain of custody, examining the laboratory’s protocol, verifying that it adhered to that protocol, and conducting confirmatory tests (including testing by the defense) can help show that the profiles really do match. Yet, “[e]rrors happen, even in the best laboratories, and even when the analyst is certain that every precaution against error was taken.”157
155. A close relative, for these purposes, would be a brother, uncle, nephew, etc. For relationships more distant than second cousins, the probability of a chance match is nearly as small as for persons of the same ethnic subgroup. Bernard Devlin & Kathryn Roeder, DNA Profiling: Statistics and Population Genetics, in 1 Modern Scientific Evidence: The Law and Science of Expert Testimony § 18–3.1.3, at 724 (David L. Faigman et al. eds., 1997) (section not included in later editions).
156. Cf. Newton E. Morton, The Forensic DNA Endgame, 37 Jurimetrics J. 477, 480 tbl. 1 (1997).
157. NRC I, supra note 6, at 89; see also Ate Kloosterman et al., Error Rates in Forensic DNA Analysis: Definition, Numbers, Impact and Communication, 12 Forensic Sci. Int’l: Genetics 775 (2014), https://doi.org/10.1016/j.fsigen.2014.04.014.
Some commentary proposes using the proportion of false positives that the particular examiner or laboratory has experienced in blind proficiency tests, or the rate of false positives on proficiency tests averaged across all laboratories, to estimate the probability of a false inclusion in the case at bar.158 Other commentators stress that there are too few proficiency tests to estimate averages accurately and question the application of an historical industry-wide error rate to a particular laboratory at a later time.159 Proponents of using averages from proficiency tests reply that this information at least can be used to provide an upper bound on the false-positive laboratory-error probability.160
Could a Close Relative Be the Source?
With enough loci to test, all individuals except some identical twins should be distinguishable. With the STR-CE technology in general use and with especially small quantities of DNA, however, this ideal is not always attainable. A thorough investigation might extend to all known relatives, but this is not always feasible, and there is always the chance that some unknown relatives are in the suspect population. Formulas are available for computing the probability that any person with a specified degree of kinship to the defendant also possesses the incriminating genotype.161 For example, the probability that an untested brother (or sister) would match at four loci (with alleles that each occur in 10% of the population) is about
158. E.g., Jonathan J. Koehler, Proficiency Tests to Estimate Error Rates in the Forensic Sciences, 12 Law, Probability & Risk 89 (2013), https://doi.org/10.1093/lpr/mgs013 (proposing blind performance tests specifically designed to estimate error rates rather than proficiency tests for quality assurance); Jonathan J. Koehler, Error and Exaggeration in the Presentation of DNA Evidence at Trial, 34 Jurimetrics J. 21, 37–38 (1993); NRC I, supra note 6, at 94 (“proficiency tests provide a measure of the false-positive and false-negative rates of a laboratory”).
159. NRC II, supra note 7, at 85–87; Devlin & Roeder, supra note 155, § 18–5.3, at 744–45. Such arguments have not persuaded the proponents of estimating the probability of error from industry-wide proficiency testing. E.g., Jonathan J. Koehler, Why DNA Likelihood Ratios Should Account for Error (Even When a National Research Council Report Says They Should Not), 37 Jurimetrics J. 425 (1997).
160. E.g., Jonathan J. Koehler, DNA Matches and Statistics: Important Questions, Surprising Answers, 76 Judicature 222, 228 (1993); Richard Lempert, After the DNA Wars: Skirmishing with NRC II, 37 Jurimetrics J. 439, 447–48, 453 (1997). For example, if there were no errors in 100 tests, a 95% confidence interval would include the possibility that the error rate could be almost as high as 3%. See NRC II, supra note 7, at 86 n.1. For an explanation of confidence intervals and the “zero-numerator problem,” see Kaye & Stern, supra note 12; see also supra section titled “What Are DNA Polymorphisms and How Are They Detected?”
161. John S. Buckleton et al., Relatedness, in Forensic DNA Evidence Interpretation 119 (John S. Buckleton et al. eds., 2d ed. 2016); Bruce S. Weir, Kinship, in Handbook of Forensic Statistics 265, 268–71 (David Banks et al. eds., 2021) (but also noting the uncertainties or complications in applying the formulas).
1/380;162 the probability that an aunt (or uncle) would match is about 1/100,000.163 With more independent loci, the probabilities will plummet.
Could an Unrelated Person Be the Source?
Another rival hypothesis is coincidence: The defendant is not the source of the crime-scene DNA but happens to have the same genotype as an unrelated individual who is the true source. In principle, this hypothesis could be addressed by genotyping everyone in the suspect population. But that is rarely feasible.164 Instead, an estimate of how frequently the incriminating genotype occurs in broad racial or ethnic populations is provided.165 The first step is to estimate the frequencies of individual alleles from samples of various populations (often referred to as reference or population databases).166 Typically, convenience samples (often from blood banks
162. For a case with conflicting calculations of the probability of an untested brother having a matching genotype, see McDaniel v. Brown, 558 U.S. 120 (2010) (per curiam). The correct computation is given in David H. Kaye, “False, but Highly Persuasive”: How Wrong Were the Probability Estimates in McDaniel v. Brown?, 108 Mich. L. Rev. First Impressions 1 (2009), https://elibrary.law.psu.edu/cgi/viewcontent.cgi?article=1059&context=fac_works; and David H. Kaye, The Interpretation of DNA Evidence: A Case Study in Probabilities, in Science Policy Decision-Making Educational Modules (Nat’l Academies of Sci., Engineering, and Med. Comm. on Preparing the Next Generation of Policy Makers for Science-Based Decisions ed. 2016), https://perma.cc/D9RM-DYDK [hereinafter Kaye, Case Study].
163. These figures follow from the equations in NRC II, supra note 7, at 113. The large discrepancy between two siblings on the one hand, and an uncle and nephew on the other, reflects the fact that the siblings have far more shared ancestry. All their genotypes are inherited through the same two parents. In contrast, a nephew and an uncle inherit from two unrelated mothers, and so will have few maternal alleles in common. As for paternal alleles, the nephew inherits not from his uncle, but from his uncle’s brother, who shares by descent only about one-half of his alleles with the uncle.
164. The suspect population normally defies enumeration, and in the typical crime where DNA evidence is found, the population of possible perpetrators is so huge that even if all of its members could be listed, they could not all be tested. As the cost of DNA profiling drops, however, it will become technically and economically feasible to have a comprehensive, population-wide DNA database that could be used to produce a list of nearly everyone whose DNA profile is consistent with the trace-evidence DNA. Whether such a system would be constitutionally and politically acceptable is another question. See David H. Kaye & Michael S. Smith, DNA Identification Databases: Legality, Legitimacy, and the Case for Population-Wide Coverage, 2003 Wis. L. Rev. 413. But the introduction of investigative genetic genealogy has renewed interest in the concept. J.W. Hazel et al., Is It Time for a Universal Genetic Forensic Database?, 362 Science 898 (2018).
165. The underlying premise is that, considering the biology of DNA markers, each possible forensic DNA genotype occurs just as often among criminal offenders as anybody else. So if we know the profile frequency in a large, relevant population group, we can use it for the unobserved set of conceivable suspects.
166. In the formative years of forensic DNA testing, defendants frequently contended that forensic databases were too small to give accurate estimates, but this argument generally proved unpersuasive. E.g., United States v. Shea, 957 F. Supp. 331, 341–43 (D.N.H. 1997); State v. Copeland, 922 P.2d 1304, 1321 (Wash. 1996). To the extent that the databases are comparable to random
or paternity cases, with the “race” of the donor being self-declared or assessed by the person collecting the sample) are used to construct the databases.167 Second, the estimated allele frequencies at each locus are combined into single-locus frequencies. The simplest computation posits an infinite population of individuals who choose their mates and reproduce independently of the forensic-identification alleles they possess.168 Then the expected population proportion of a pair of alleles (a single-locus genotype) is essentially the product of allele frequencies.169 Finally, the single-locus frequencies are multiplied together.170
Because the frequencies vary across census groups (White, Black, Hispanic, Asian, and Native American), it became common to present separate estimates for these groups, at least in cases in which the race of the source of the trace evidence was unknown. For example, if a woman was abducted from a rest stop on an interstate highway and sexually assaulted, the suspect population could include people of all census categories.171 Random-match probabilities for all the major
samples, confidence intervals can be used to indicate the uncertainty related to sample size. Unfortunately, the meaning of a confidence interval is subtle, and the estimate commonly is misconstrued. See Kaye & Stern, supra note 12.
167. A few experts have testified that no meaningful conclusions can be drawn in the absence of random sampling. E.g., People v. Soto, 88 Cal. Rptr. 2d 34 (1999); State v. Anderson, 881 P.2d 29, 39 (N.M. 1994). The 1996 NRC report suggests that for the purpose of estimating allele frequencies, convenience sampling should give results comparable to random sampling, and it discusses procedures for estimating the random sampling error. NRC II, supra note 7, at 126–27, 146–48, 186. The courts generally have rejected the argument that random samples are essential to valid or generally accepted random-match probabilities. See section titled “A Brief History of DNA Evidence” above.
168. Of course, people do not choose their mates by a lottery. “Random mating” simply indicates that the choices are uncorrelated with the specific alleles that make up the genotypes in question. A further condition of the random-mating model is that there is no systematic relationship between the particular alleles that an offspring receives and the chance that the child will transmit an allele to the next generation.
169. If the single-locus genotype is homozygous (for example, the allele from each parent is A1), then expected proportion is just p1 × p1, or p12, where p1 is the proportion of all alleles in the population that are type A1. If the pair of alleles at a locus are distinct (call them A1 and A2), then the single-locus heterozygous genotype proportion is 2p1p2. For example, suppose that 10% of the sperm in the gene pool of the population carry allele 1, and 50% carry allele 2. Similarly, 10% of the eggs carry A1, and 50% carry A2. (Other sperm and eggs carry other types.) With random mating, we expect 10% × 10% = 1% of all the fertilized eggs to be A1A1, and another 50% × 50% = 25% to be A2A2. These constitute two distinct homozygote profiles. Likewise, we expect 10% × 50% = 5% of the fertilized eggs to be A1A2 and another 50% × 10% = 5% to be A2A1. These two configurations produce indistinguishable profiles—a peak, band, or dot for A1 and another mark for A2. So the expected proportion of heterozygotes is 5% + 5% = 10%.
These proportions are known as Hardy-Weinberg proportions. Even if two populations with distinct allele frequencies are thrown together, within the limits of chance variation, random mating produces Hardy-Weinberg equilibrium in a single generation.
170. When the various single-locus genotypes are statistically independent of one another, the population is said to be in linkage equilibrium.
171. United States v. Jakobetz, 955 F.2d 786 (2d Cir. 1992).
groups enable the jury to see how much the estimates vary for different parts of the population even if they cannot determine which of these parts is most salient.
In cases in which the suspect population seems limited to a single major population group—for example, where the victim is sure that the assailant looked Hispanic and spoke with a Spanish accent—only the frequency estimated for that group might be presented.172 But what if the victim is mistaken or the perpetrator is of mixed ancestry? If the wrong racial or ethnic database were used to estimate a profile frequency, it would yield too small (or too large) a random-match probability. Thus, even in cases in which the evidence points to a subpopulation, there is an argument for giving the estimates for several groups.173
A concern often raised with racial classifications involving DNA evidence is that references to race and DNA genotypes of any kind will “reify” the idea that racial categories are fundamentally biological rather than socially constructed.174 There are ways to avoid the census-type labels. Allele frequency data can be collected from regions across the globe, and the spectrum of resulting profile frequency estimates could be presented in every case.175 Alternatively, only the highest frequency estimate across this spectrum could be selected to supply a “conservative” estimate (that is, an overestimate). One might even create an unrealistic population by picking, for each and every allele, the frequency that is the largest in any of the groups and combining those maximum frequencies.176 Ideas like these have been proposed,177 and
172. Cf. People v. Pizarro, 12 Cal. Rptr. 2d 436, 441 (Ct. App. 1992), after remand, 3 Cal. Rptr. 3d 21 (Ct. App. 2003), review denied (Oct. 15, 2003).
173. David H. Kaye, The Role of Race in DNA Evidence: What Experts Say, What California Courts Allow, 37 Sw. U. L. Rev. 303 (2008).
174. E.g., Troy Duster, Race and Reification in Science, 307 Science 1050 (2005); section titled “Biogeographic Ancestry Testing” below.
175. The FBI’s STR allele-frequency database has samples from “African Americans, Caucasians, Southeast Hispanics, Southwestern Hispanics, Bahamians, Jamaicans, Trinidadians, Apaches, Navajos, Chamorros and Filipinos.” Tamyra R. Moretti et al., Population Data on the Expanded CODIS Core STR Loci for Eleven Populations of Significance for Forensic DNA Analyses in the United States, 25 Forensic Sci. Int’l: Genetics 175, 175 (2016), https://doi.org/10.1016/j.fsigen.2016.07.022.
176. This is a rough statement of one of the controversial “ceiling” methods “strongly recommend[ed]” in NRC I, supra note 6, at 82–85.
177. An advisory commission appointed by the Department of Justice noted that a more refined population-genetics model (mentioned below) could be used to treat the entire United States as a single, structured population, obviating the reporting of estimates by more specific groups. Nat’l Comm’n on the Future of DNA Evidence, The Future of DNA Evidence: Predictions of the Research and Development Working Group 5 (2000); see also Robert F. Oldt & Sreetharan Kanthaswamy, Expanded CODIS STR Allele Frequencies—Evidence for the Irrelevance of Race-based DNA Databases, 42 Legal Med. 101642 (2020), https://doi.org/10.1016/j.legalmed.2019.101642 (questioning the value of racial labels when typing at 20 STR loci is successful). Another procedure that avoids racial categories is simply to give the (larger) probability of a random match to a brother or sister at the loci used in the testing. Because full siblings always are more closely related than are
some were used in cases.178 However, the dominant reason given for looking to more narrowly defined populations was a perception that the model of randomly mating major racial populations was just too simple. Perhaps linguistic, religious, or other subgroups within the broader populations mate almost exclusively among themselves. If these subpopulations happen to have varying allele frequencies, then the estimates for the DNA-genotype frequencies in the broader groups could understate (or overstate) the true frequencies for the racial groups or be inapposite in a case in which the suspect population is confined to a narrow subgroup.
For a time, the likely magnitude of the effect of this “population structure” on profile-frequency estimates was hotly contested.179 Today, a quantity denoted as FST or θ (theta)180 often is used in formulas designed to account for population structure;181 however, there is a continuing discussion of what value to use for θ,182 and some researchers have developed further refinements.183
Frequencies, Probabilities, and Prejudice
Up to this point, we have described the estimates for genotype frequencies (often presented as random-match probabilities) that commonly are quoted in conjunction with the finding that the trace-evidence sample contains DNA of the same
randomly drawn pairs from even a narrowly defined subpopulation, this “Sib Method . . . provides a rough upper limit for the actual match probability.” Nat’l Comm’n, supra, at 5.
178. E.g., Brim v. State, 695 So.2d 268 (Fla. 1997).
179. Jay D. Aronson, Genetic Witness: Science, Law, and Controversy in the Making of DNA Profiling (2007); Kaye, supra note 1, at 121–26. By the mid-1990s, the population-structure objection to admitting random-match probabilities had lost its punch. See, e.g., Kaye, supra note 1; supra section titled “A Brief History of DNA Evidence.”
180. See David Balding & Richard A. Nichols, DNA Profile Match Probability Calculation: How to Allow for Population Stratification, Relatedness, Database Selection and Single Bands, 64 Forensic Sci. Int’l 125 (1994); Gill et al., supra note 42, at 29–38.
181. The recommendations of the 1996 NAS committee for computing random-match probabilities with these formulas for broad populations and particular subpopulations are summarized in the second edition of this guide. See Reference Manual on Scientific Evidence (2d ed. 2000).
182. See John Buckleton et al., Population-specific FST Values for Forensic STR Markers: A Worldwide Survey, 23 Forensic Sci. Int’l: Genetics 91 (2016); Bruce S. Weir, DNA Frequencies and Probabilities, in Handbook of Forensic Statistics 251, 256–57 (David Banks et al. eds., 2021); Weir, supra note 161, at 266–68. The U.K. Forensic Science Regulator recommends the value of 0.03 as “sufficiently conservative that it is almost certainly favourable to defendants even allowing for alternative contributors to come from very different ethnic populations.” Forensic Science Regulator (U.K.), Guidance: Allele Frequency Databases and Reporting Guidance for the DNA (Short Tandem Repeat) Profiling, FSR-G-213 Issue 2, § 14.1.4 (2020); however, “in unusual cases involving small and isolated populations that may be highly differentiated from available databases,” 0.05 could be used. Id. § 14.1.1.
183. See Mark A. Jobling, Forensic Genetics Through the Lens of Lewontin: Population Structure, Ancestry and Race, 377 Phil. Transactions Royal Soc’y B 20200422 (2022) (citing three papers).
type as the defendant’s. Assuming that the statistical methods meet Daubert’s demand for scientific validity and reliability, and thus satisfy Federal Rule of Evidence 702 (or, in some states, Frye’s requirement of general acceptance in the scientific community), a further issue can arise under Rule 403: To what extent will the presentation assist the jury in understanding the meaning of a match so that the jury can give the evidence the weight that it deserves? This question involves psychology and law, and we summarize the arguments about probative value and prejudice that have been made in litigation and in the legal and scientific literature. How the legal issue of the admissibility of any particular statistic generally should be resolved under the balancing standard of Rule 403 may turn not only on the general features of the evidence described here, but on the context and circumstances of particular cases.
Are Frequencies or Probabilities Prejudicial Because They Are So Small?
The most common form of expert testimony about matching DNA involves an explanation of how the laboratory ascertained that the defendant’s DNA has the profile of the trace-evidence sample plus an estimate of the profile frequency or random-match probability.184 It has been suggested, however, that jurors do not understand probabilities in general, and that infinitesimal match probabilities will so bedazzle jurors that they will not appreciate the other evidence in the case or any innocent explanations for the match.185 Empirical research into this hypothesis does not clearly support the argument that jurors will overweight the probability.186 The details of how the probability is presented and countered may be important, and remedies short of exclusion are available.187 The practice in
184. A more flexible alternative that has widespread support among forensic DNA scientists and forensic statisticians is the likelihood ratio or Bayes’ factor discussed infra section titled “Mixtures.”
185. Cf. Gov’t of the Virgin Islands v. Byers, 941 F. Supp. 513, 527 (D.V.I. 1996) (“Vanishingly small probabilities of a random match may tend to establish guilt in the minds of jurors and are particularly suspect.”).
186. Thomas Busey et al., Validating Strength-of-Support Conclusion Scales for Fingerprint, Footwear, and Toolmark Impressions, 67 J. Forensic Sci. 936 (2022) (“whether jurors can understand more complex statistical terms such as likelihood ratios and random match probabilities is an empirical question with no clear answer in the literature”); see generally Kristy A. Martire, How Well Do Lay People Comprehend Statistical Statements from Forensic Scientists, in Handbook of Forensic Statistics 201 (David Banks et al. eds., 2021); David H. Kaye et al., Statistics in the Jury Box: Do Jurors Understand Mitochondrial DNA Match Probabilities?, 4 J. Empirical Legal Stud. 797 (2007).
187. United States v. Graves, 465 F. Supp. 2d 450, 458 (E.D. Pa. 2006) (“with cross-examination, proper explanations, and clarifying jury instructions, the probative value of the sneaker evidence [with RMPs of about 1/3,000] is not substantially outweighed by the danger of unfair prejudice and confusion”); United States v. Santiago, 156 F. Supp. 2d 145, 151 (D.P.R.
the United Kingdom is to cap reported random-match probabilities at “1 in 1 billion.”188
Thus, although there once was a line of cases that excluded probability testimony in criminal matters, by the mid-1990s, no jurisdiction excluded DNA match probabilities on this basis.189 The opposite argument—that relatively large random-match probabilities are prejudicial because jurors will not appreciate that they make the DNA match unimpressive—also has been advanced without much success.190
Are Frequencies or Probabilities Prejudicial Because They Might Be Transposed?
A related concern is that the jury will misconstrue the random-match probability as the probability that the evidence DNA came from a random individual. The words are almost identical, but the probabilities can be different. The random-match probability is the probability that the suspect’s DNA matches the trace-evidence sample calculated on the proposition that the suspect is not the true source of that sample (and is not a close relative). The proposition is a hypothesis about who the source really is; it asserts that the true source is someone other than the suspect. But the laboratory cannot evaluate the probability of this hypothesis on the basis of
2001); NRC II, supra note 7, at 197 (suitable cross-examination, defense experts, and jury instructions might reduce the risk of an unwarranted sense of certainty).
188. Forensic Science Regulator, Guidance: Allele Frequency Databases and Reporting Guidance for the DNA (Short Tandem Repeat) Profiling (2014) (FSR-G-213 Issue 1, Recommendation 4, retained in Issue 2, 2020, §§ 8 & 10). In part, this limit reflects the concern that the assumptions of population genetics and statistical models do not hold exactly, reducing the probative value of much smaller numbers. It also is in the spirit of the 2023 amendment to Rule 702 intended “to emphasize that . . . [a] testifying expert’s opinion must stay within the bounds of what can be concluded by a reliable application of the expert’s basis and methodology.” Fed. R. Evid. 702 advisory committee’s note to the 2023 amendment. But see Tacha Hicks et al., A Logical Framework for Forensic DNA Interpretation, 13 Genes 957 (2022), https://doi.org/10.3390/genes13060957 (Noting that when “the Forensic Science Service introduced the ten locus STR system into casework,” the one-in-a-billion cap was used as “a temporary expedient” because it was “argued that the extent of investigations into the independence assumptions . . . was insufficient to justify the robustness of LRs in excess of one billion”; yet “to this day, there still seems to be little in the way of large-scale between-locus dependence effects,” making the practice for the much larger “number of loci now in routine use . . . a peculiar state of affairs.”).
189. E.g., United States v. Chischilly, 30 F.3d 1144 (9th Cir. 1994); State v. Weeks, 891 P.2d 477, 489 (Mont. 1995); see also State v. Bauldwin, 811 N.W.2d 267, 288 (Neb. 2012).
190. E.g., United States v. McCluskey, 954 F. Supp. 2d 1224, 1273–75 (D.N.M. 2013); State v. Tucker, 920 N.W.2d 680, 688–89 (Neb. 2018). But see United States v. Graves, supra note 187, at 459 (“even with appropriate safeguards, the minimal probative value of the umbrella DNA evidence—in which half of the relevant population cannot be excluded as a contributor to the DNA sample—is substantially outweighed by the danger of unfair prejudice and confusion of the issues”).
the DNA evidence itself. It can only provide the probability of an event such as the suspect’s DNA profile matching the crime-scene DNA profile if the true source is genetically unrelated to the suspect (or related in a specified manner). The occurrence of the match certainly is circumstantial evidence in favor of the hypothesis that the suspect is the source; however, automatically “equating source probability with random match probability” is “faulty reasoning.”191
Sliding from the probability of the circumstantial evidence to the probability of the circumstances themselves (the hypotheses about the evidence) often is called the prosecutor’s fallacy,192 although instances are hardly confined to prosecutors.193 Statisticians know it as the fallacy of the transposed conditional.194 Despite the attention the transposition fallacy has received, no federal court has excluded a random-match probability as unfairly prejudicial simply because the jury might misinterpret it as a probability that the defendant is the
191. McDaniel v. Brown, 558 U.S. 120, 128 (2010) (per curiam).
192. Id. In Brown, a DNA analyst, at the prompting of the prosecution, suggested that a random-match probability of 1/3,000,000 implied a 0.000033 probability that the defendant was not the source of the DNA found on the victim’s clothing. The Court unanimously held that a federal writ of habeas corpus should not have been issued in light of the limitations on federal review of state convictions. The prisoner argued, inter alia, that the transposed conditional probability made the DNA evidence so unreliable that its use deprived him of due process. The Court did not reach this issue, as it had not been raised below. The probabilities associated with the DNA evidence in the case are discussed in detail in Kaye, Case Study, supra note 162.
193. It also appears in statements from defense counsel, scientists, journalists, and judges. E.g., Williams v. Bauman, 759 F.3d 630, 632 (6th Cir. 2014) (“a 1–in–7.663 quadrillion chance that it came from someone else”); United States v. Ford, 683 F.3d 761, 768 (7th Cir. 2012) (“[t]he probability that the DNA was someone else’s would be 7 percent if the comparison were confined to the first location”); see also David H. Kaye et al., The New Wigmore, A Treatise on Evidence: Expert Evidence § 14.1.2(a) (2d ed. 2011) (chapter not included in the current edition) (collecting opinions expressing the fallacy); 2 Paul C. Giannelli et al., Scientific Evidence § 18.04[3][b][4], at 1893 n.384 (“nearly every appellate decision in the U.S. on the sufficiency of a DNA cold hit has fallen victim to the fallacy”).
There is an opposing “defendant’s fallacy” of dismissing or undervaluing matches because other matches are to be expected in unrealistically large populations of potential suspects. For example, defense counsel might argue that (1) with a random-match probability of one in a million, we would expect to find three or four unrelated people with the requisite genotypes in a major metropolitan area with a population of 3.6 million; (2) the defendant just happens to be one of these three or four, which means that the chances are at least 2 out of 3 that someone unrelated to the defendant is the source; so (3) the DNA evidence does nothing to incriminate the defendant. The problem with this argument is that in a case involving both DNA and non-DNA evidence against the defendant, it is unrealistic to assume that there are 3.6 million equally likely suspects. When juries are confronted with both fallacies, the defendant’s fallacy seems to dominate. NRC II, supra note 7, at 198; cf. Jonathan J. Koehler, The Psychology of Numbers in the Courtroom: How to Make DNA-Match Statistics Seem Impressive or Insufficient, 74 S. Cal. L. Rev. 1275 (2001) (discussing ways of framing the evidence that make it more or less persuasive).
194. See Kaye & Stern, supra note 12, app’x section C (describing the fallacy and the relationship between the conditional probabilities); Ian W. Evett, Avoiding the Transposed Conditional, 35 Sci. & Just. 127 (1995).
source of the forensic DNA.195 Courts, however, have noted the need to have the concept “properly explained,”196 and prosecutorial or expert misrepresentations of the random-match probabilities for DNA and other trace evidence have produced reversals or contributed to the setting aside of verdicts.197
Are Random-Match Probabilities That Are Smaller Than False-Positive Error Probabilities Irrelevant or Prejudicial?
Some scientists and lawyers have maintained that match probabilities are logically irrelevant when they are far smaller than the probability of a frame-up, a blunder in labeling samples, cross-contamination, or other events that would
195. See, e.g., United States v. McCluskey, 954 F. Supp. 2d 1224, 1291 (D.N.M. 2013); United States v. Morrow, 374 F. Supp. 2d 51, 66 (D.D.C. 2005) (“careful oversight by the district court and proper explanation can easily thwart this issue”).
196. United States v. Shea, 957 F. Supp. 331, 345 (D.N.H. 1997); see also United States v. Chischilly, 30 F.3d 1144, 1158 (9th Cir. 1994) (stating that the government must be “careful to frame the DNA profiling statistics presented at trial as the probability of a random match, not the probability of the defendant’s innocence that is the crux of the prosecutor’s fallacy”). The 1996 NRC committee suggested that “if the initial presentation of the probability figure, cross-examination, and opposing testimony all fail to clarify the point, the judge can counter [the fallacy] by appropriate instructions to the jurors that minimize the possibility of cognitive errors.” NRC II, supra note 7, at 198 (footnote omitted). The committee formulated the following instruction to define the random-match probability:
In evaluating the expert testimony on the DNA evidence, you were presented with a number indicating the probability that another individual drawn at random from the [specify] population would coincidentally have the same DNA profile as the [bloodstain, semen stain, etc.]. That number, which assumes that no sample mishandling or laboratory error occurred, indicates how distinctive the DNA profile is. It does not by itself tell you the probability that the defendant is innocent. Id. at 198 n.93. An alternative adopted in England is to confine the prosecution to stating a frequency rather than a probability. See Kaye et al., supra note 193, § 14.1.2(b); cf. D.H. Kaye, The Admissibility of “Probability Evidence” in Criminal Trials—Part II, 27 Jurimetrics J. 160, 168 (1987) (similar proposal).
197. E.g., United States v. Massey, 594 F.2d 676, 681 (8th Cir. 1979) (explaining that in closing argument about hair evidence, “the prosecutor ‘confuse[d] the probability of concurrence of the identifying marks with the probability of mistaken identification’”) (alteration in original); cf. Whack v. State, 73 A.3d 186, 198 (Md. 2013) (reversing defendant’s conviction because of the prosecution’s closing argument about two probabilities and admonishing that “counsel have a responsibility to take extra care in describing DNA evidence, particularly when it comes to statistical probabilities”); State v. Phillips, 844 S.E.2d 651, 658 (S.C. 2020) (reversing partly as a result of inadequacies and errors in the presentation of random-match probabilities for “touch” DNA evidence and noting that “even when the concepts of touch DNA, non-exclusion DNA, and random match probability are completely and accurately presented to a jury, there is significant potential the testimony will be confusing and misleading”).
yield a false positive.198 The argument is that the jury should concern itself only with the chance that the forensic sample is reported to match the defendant’s profile even though the defendant is not the source. Match probabilities address only one path to such a false-positive report—a coincidence in correctly ascertained profiles. They do not consider the probability of a false-positive report because of fraud or an error in the collection, handling, or analysis of the DNA samples. Unless those probabilities are essentially zero, the match probability understates the chance of a reported match arising when DNA from a nonsource is compared to DNA from the source of the trace-evidence sample.
This mathematical observation has led to arguments that because probability of these other possible explanations for a match are larger than the very small random-match probabilities for most STR profiles, the latter probabilities are irrelevant. Commentators have crafted theoretical, doctrinal, and practical rejoinders to this claim.199 The essence of the counterargument is that it is logical to give jurors information about kinship or random-match probabilities because, even if these numbers do not give the whole picture, they address pertinent hypotheses about the true source of the trace evidence.
It also has been argued that even if very small match probabilities are logically relevant, they are unfairly prejudicial in that they will cause jurors to neglect the probability of a match arising because of a false-positive laboratory error.200 A court that shares this concern might require the expert who presents a random-match probability also to report a probability that the laboratory is mistaken about
198. E.g., Jonathan J. Koehler et al., The Random Match Probability in DNA Evidence: Irrelevant and Prejudicial?, 35 Jurimetrics J. 201 (1995); Richard C. Lewontin & Daniel L. Hartl, Population Genetics in Forensic DNA Typing, 254 Science 1745, 1749 (1991), https://doi.org/10.1126/science.1845040 (“[p]robability estimates like 1 in 738,000,000,000,000 . . . are terribly misleading because the rate of laboratory error is not taken into account”).
199. See Kaye et al., supra note 193, § 14.1.1 (discussing the issue).
200. Some commentators believe that this prejudice is so likely and so serious that “jurors ordinarily should receive only the laboratory’s false positive rate. . . .” Richard Lempert, Some Caveats Concerning DNA as Criminal Identification Evidence: With Thanks to the Reverend Bayes, 13 Cardozo L. Rev. 303, 325 (1991) (emphasis added). The 1996 NRC committee was skeptical of this view, especially when the defendant has had a meaningful opportunity to retest the DNA at a laboratory of his or her choice, and it suggested that judicial instructions can be crafted to avoid this form of prejudice. NRC II, supra note 7, at 199. Pertinent psychological research includes Dale A. Nance & Scott B. Morris, Juror Understanding of DNA Evidence: An Empirical Assessment of Presentation Formats for Trace Evidence with a Relatively Small Random Match Probability, 34 J. Legal Stud. 395 (2005); Dale A. Nance & Scott B. Morris, An Empirical Assessment of Presentation Formats for Trace Evidence with a Relatively Large and Quantifiable Random Match Probability, 42 Jurimetrics J. 403 (2002); Jason Schklar & Shari Seidman Diamond, Juror Reactions to DNA Evidence: Errors and Expectancies, 23 Law & Hum. Behav. 159, 179 (1999), https://doi.org/10.1023/A:1022368801333 (concluding that separate figures for laboratory error and a random match to a correctly ascertained profile are desirable in that “[j]urors . . . may need to know the disaggregated elements that influence the aggregated estimate as well as how they were combined in order to evaluate the DNA test results in the context of their background beliefs and the other evidence introduced at trial”).
the profiles. Of course, for reasons given in the section titled “How Are Samples Created and Handled?,” above, some experts would deny that they can provide a meaningful statistic for the case at hand, but they could report the results of proficiency tests and leave it to the jury to use this figure as best it can in considering whether a false-positive error has occurred.201 In any event, the courts have been unreceptive to efforts to replace random-match probabilities with a blended figure that incorporates the risk of a false-positive error202 or to exclude random-match probabilities that are not accompanied by a separate false-positive error probability.203
“Rarity,” Source, and Uniqueness Testimony
Having surveyed the issues related to the value and dangers of probabilities and statistics for DNA evidence, we turn to a related issue that can arise under Rules 702 and 403: Should an expert be permitted to offer a nonnumerical judgment about the DNA profiles? Many courts have held that a DNA match is inadmissible unless the expert attaches a scientifically valid number to the match. Indeed, some opinions state that this requirement flows from the nature of science
201. Cf. Williams v. State, 679 A.2d 1106, 1120 (Md. 1996) (reversing because the trial court restricted cross-examination about the results of proficiency tests involving other DNA analysts at the same laboratory). But see United States v. Shea, 957 F. Supp. 331, 344 n.42 (D.N.H. 1997) (“The parties assume that error rate information is admissible at trial. This assumption may well be incorrect. Even though a laboratory or industry error rate may be logically relevant, a strong argument can be made that such evidence is barred by Fed. R. Evid. 404 because it is inadmissible propensity evidence.”).
202. United States v. McCluskey, 954 F. Supp. 2d 1224, 1270–73 (D.N.M. 2013); United States v. Ewell, 252 F. Supp. 2d 104, 113–14 (D.N.J. 2003); United States v. Shea, 957 F. Supp. 331, 334–45 (D.N.H. 1997); People v. Reeves, 109 Cal. Rptr. 2d 728, 753 (Ct. App. 2001); State v. Tester, 968 A.2d 895 (Vt. 2009); cf. Armstead v. State, 673 A.2d 221, 245 (Md. 1996) (the failure to combine a random-match probability with an error rate on proficiency tests that was many orders of magnitude greater (and that was placed before the jury) did not deprive the defendant of due process). Formulas for incorporating error into the likelihood are given in Tacha Hicks et al., A Framework for Interpreting Evidence, in Forensic DNA Evidence Interpretation 37, 73 (John Buckleton et al. eds., 2d ed. 2016), https://doi.org/10.1201/b19680-3.
203. McCluskey, 954 F. Supp. 2d at 1270–73; United States v. Trala, 162 F. Supp. 2d 336, 350–51 (D. Del. 2001); United States v. Lowe, 954 F. Supp. 401, 415–16 (D. Mass. 1997), aff’d, 145 F.3d 45 (1st Cir. 1998) (a “theoretical” error rate need not be presented when quality-assurance standards have been followed and defendant had the opportunity to retest the sample); Roberts v. United States, 916 A.2d 922, 930–31 (D.C. 2007); Roberson v. State, 16 S.W.3d 156, 168 (Tex. Crim. App. 2000) (error rate not needed when laboratory was accredited and underwent blind proficiency testing); Tester, 968 A.2d 895 (finding when the laboratory chemist stated that “[t]here is no error rate to report” because the number of proficiency trials was insufficient, the random-match probability was admissible and preferable to presenting the finding of a match with no accompanying statistic).
itself.204 However, this view has been challenged,205 and not all courts agree that an expert must explain the power of a DNA match in purely numerical terms.206
Instead of presenting numerical frequencies or match probabilities, a scientist could characterize a multilocus STR profile as “rare,” “extremely rare,” or the like.207 A few opinions suggest that a purely qualitative presentation would be admissible,208 but with the availability of statistically sound estimates of frequencies or conditional probabilities, such testimony is itself quite rare. When numerical estimates are possible, it is not clear what the expert’s verbal tag accomplishes.
The most extreme case of a purely verbal description of the infrequency of a profile occurs when that profile can be said to be unique. Of course, the uniqueness of any object, from a snowflake to a fingerprint, in a population that cannot be enumerated, never can be proved directly. As with all sample evidence, one must generalize from the sample to the entire population. There is always some probability that a census would prove the generalization to be false. Almost three decades ago, the second National Research Council (NRC) committee therefore wrote:
[T]here is no “bright-line” standard in law or science that can pick out exactly how small the probability of the existence of a given profile in more than one member of a population must be before assertions of uniqueness are justified. . . . There might already be cases in which it is defensible for an expert to assert that, assuming that there has been no sample mishandling or laboratory error, the profile’s probable uniqueness means that the two DNA samples come from the same person.209
204. E.g., State v. Cauthron, 846 P.2d 502 (Wash. 1993).
205. See, e.g., State v. Hummert, 933 P.2d 1187, 1191 (1997) (“We believe this view to be seriously incorrect.”); Commonwealth v. Crews, 640 A.2d 395, 402 (Pa. 1994) (“[T]he factual evidence of the physical testing of the DNA samples and the matching alleles, even without statistical conclusions, tended to make appellant’s presence more likely than it would have been without the evidence, and was therefore relevant.”). The National Research Council committee wrote that science only demands “underlying data that permit some reasonable estimate of how rare the matching characteristics actually are,” and “[o]nce science has established that a methodology has some individualizing power, the legal system must determine whether and how best to import that technology into the trial process.” NRC II, supra note 7, at 192.
206. E.g., Rodriguez v. State, 273 P.3d 845, 851 (Nev. 2012); People v. Her, 157 Cal. Rptr. 3d 40 (Cal. Ct. App. 2013). For discussion of pure “defendant-not-excluded” testimony, see United States v. Morrow, 374 F. Supp. 2d 51 (D.D.C. 2005); Kaye et al., supra note 193, § 15.4.
207. Banks v. State, 219 So.3d 19, 28 (Fla. 2017) (“by the time you get to 13 [STR loci] it becomes extremely rare, extremely”); cf. State v. Hummert, 933 P.2d 1187, 1193 (Ariz. 1997) (testimony that “of their own experience, they believed such a [three-locus RFLP-VNTR] random match would be very uncommon” was admissible).
208. State v. Bloom, 516 N.W.2d 159, 166–67 (Minn. 1994) (“Since it may be pointless to expect ever to reach a consensus on how to estimate, with any degree of precision, the probability of a random match and that, given the great difficulty in educating the jury as to precisely what that figure means and does not mean, it might make sense to simply try to arrive at a fair way of explaining the significance of the match in a verbal, qualitative, nonquantitative, nonstatistical way.”).
209. NRC II, supra note 7, at 194.
Before concluding that a DNA profile is unique in a given population, however, a careful expert also should consider not only the random-match probability (which pertains to unrelated individuals) but also the chance of a match to a close relative. Indeed, the possible existence of an unknown, identical twin also means that a scientist never can be absolutely certain that crime-scene evidence could have come from only the defendant.
Courts have accepted or approved of expert assertions of uniqueness or of individual-source identification.210 For these assertions to be justified, a large number of sufficiently polymorphic loci must have been tested, making the probabilities of matches to both relatives and unrelated individuals so tiny that the probability of finding another person who could be the source within the relevant population is negligible.211 But deciding what probability is negligible is a personal or policy judgment rather than an objective fact, and trusting the probability models at the extremes where they cannot be experimentally tested
210. E.g., United States v. Carney, No. 1:20-cr-121, 2022 WL 1155902, at *1 (S.D. Ohio Apr. 19, 2022) (“in the absence of an identical twin, Furious Carney is the source of the major DNA profile”); United States v. Davis, 602 F. Supp. 2d 658 (D. Md. 2009) (“the random match probability figures . . . are sufficiently low so that the profile can be considered unique”); People v. Cordova, 358 P.3d 518, 538 (2015) (because “the odds were astronomical,” an “opinion that defendant was the source of the evidentiary samples to a reasonable scientific certainty was reasonable”); State v. Hauge, 79 P.3d 131 (Haw. 2003) (uniqueness); Young v. State, 879 A.2d 44, 46 (Md. 2005) (holding that “when a DNA method analyzes genetic markers at sufficient locations to arrive at an infinitesimal random match probability, expert opinion testimony of a match and of the source of the DNA evidence is admissible”; hence, it was permissible to introduce a report providing no statistics but stating that “(in the absence of an identical twin), Anthony Young (K1) is the source of the DNA obtained from the sperm fraction of the Anal Swab (R1)”); State v. Buckner, 941 P.2d 667, 668 (Wash. 1997) (in light of 1996 NRC Report, “we now conclude there should be no bar to an expert giving his or her expert opinion that, based upon an exceedingly small probability of a defendant’s DNA profile matching that of another in a random human population, the profile is unique”).
211. Three distinct probabilities arise in speaking of the uniqueness of DNA profiles. First, there is the probability of a match to a single, randomly selected individual in the population. This is the random-match probability. Second, there is the probability that the particular profile is unique. This probability involves pairing the profile with every member of the population. Third, there is the probability that all pairs of all profiles are unique. The first probability is larger than the second, which is many times larger than the third. Uniqueness or source testimony need only establish that the one DNA profile in the trace evidence is unique—and not that all DNA profiles are unique. Thus, it is the second probability, properly computed, that must be quite small to warrant the conclusion that no one but the defendant (and any identical twins) could be the source of the crime-scene DNA. See David H. Kaye, Identification, Individuality, and Uniqueness: What’s the Difference?, 8 Law, Probability & Risk 85 (2009), https://doi.org/10.1093/lpr/mgp018.
Formulas for estimating all these probabilities are given in NRC II, supra note 7, but DNA analysts and judges sometimes infer uniqueness on the basis of incorrect intuitions about the size of the random-match probability. See David J. Balding, Weight-of-Evidence for Forensic DNA Profiles 148 (2005) (describing “the uniqueness fallacy”); cf. State v. Lee, 976 So. 2d 109, 117 (La. 2008) (incorrect but harmless miscalculation).
makes some scientists wary of claiming uniqueness and making source attributions on the basis of DNA evidence.212
Special Issues in Human DNA Testing
Y Chromosomes
Genetic Principles
To understand how Y chromosomes are used in forensic-DNA science, we need to recall a few principles of the genetics of sexual reproduction mentioned in the section above titled “Sexual Reproduction and the Genome.” A basic point is that a male child receives an X chromosome from the genetic mother and a Y from the genetic father,213 whereas female children receive two X chromosomes, one from each parent.214 Thus, genetic males are type XY and genetic females are XX.215
A second fundamental point is that every sex cell (an egg or a sperm) has only one copy of each parental chromosome, selected at random from the pairs carried by each parent. Hence, about half the sperm cells in a genetic male have a Y chromosome, whereas the male’s bodily (somatic) cells all have the full XY pair of chromosomes.
Third—and this is crucial in forensic identification with Y chromosomes—when a sperm cell forms, there is limited recombination (exchanges of segments) between X and Y chromosomes. Along most of its length, the Y chromosome stays intact, and only occasional intergenerational mutations produce diversity among Y chromosomes within a population. In other words, the Y loci used in forensic identification are linked rather than independent. They are inherited as a single block—a haplotype—from father to son, and all the men in the same paternal line (up to the last mutation giving rise to a new line in the family tree) would match the trace-evidence sample’s Y haplotype. At the same time, men from a different paternal lineage (one with no recent male ancestor in common) might have developed the same haplotype (depending on the history of mutations
212. John S. Buckleton et al., Single Source Samples, in Forensic DNA Evidence Interpretation 203, 207–15 (John S. Buckleton et al. eds., 2d ed. 2016).
213. All told, the Y chromosome represents about 2% of the total human genome and is much smaller than its X counterpart.
214. This pattern is a consequence of the fact that certain genes in the Y chromosome result in development as a male rather as than a female.
215. Sometimes, progeny are born with fewer or more than the usual two sex chromosomes. Jeannie Visootsak & John M. Graham, Jr., Klinefelter Syndrome and Other Sex Chromosomal Aneuploidies, 1 Orphanet J. Rare Diseases 42 (2006), https://doi.org/10.1186/1750-1172-1-42; Brittany Sood & Roselyn W. Clemente Fuentes, Jacobs Syndrome, StatPearls (2022), https://perma.cc/62WT-W4S3; supra note 22.
giving rise to each line). If so, men from both lines would match a trace-evidence sample with the shared haplotype.
Like the other 22 paired chromosomes (the autosomes), the Y chromosome contains not only genes but also STRs, SNPs, and other parts that vary from one person (or group of people) to the next. A particular haplotype is thus some combination of the variants at some of these loci.
Forensic Value
In directly comparing DNA profiles, there normally is not much point in adding Y-STRs to the autosomal profile. The rarity of the multilocus autosomal STR genotypes already makes them very discriminating.216 Nevertheless, the patrilineal inheritance of Y chromosomes can be useful for determining the number of male contributors to a mixed semen sample in sexual assault cases; for discerning which male haplotype is present when a genetic male’s autosomal STR alleles cannot be detected in the midst of a much larger quantity of female DNA;217 for familial searching in criminal DNA databases;218 for inferring biogeographical ancestry as an investigative lead;219 and for establishing kinship in immigration cases or other situations.220 A famous example of the last application is the Y-chromosome testing of members of the acknowledged families of President Thomas Jefferson and Sally Hemings that revealed Sally’s last child had a fairly rare Y haplotype that President Jefferson—and everyone else in the Jefferson male clan—possessed.221
216. Moreover, some geneticists are not confident that Y-STRs are independent of autosomal STRs, making it unclear how to arrive at an estimate for a profile frequency or probability. See Weir, supra note 182, at 258 (“[A]lthough the dependencies [among autosomal STRs and Y-chromosome and mitochondrial systems] are low, they do exist. Combining match probability estimates over systems is not generally recommended.”).
217. E.g., State v. Jones, 345 P.3d 1195 (Utah 2015) (DNA on fingernail clippings from female homicide victim who struggled with her attacker); State v. Maestas, 299 P.3d 892 (Utah 2012) (same).
218. See infra section titled “Kinship trawling.”
219. See infra section titled “Biogeographic ancestry testing.”
220. For example, to help identify human remains as those of a male reported as missing, DNA from the remains might be compared to a sample from a father, brother, uncle, or other relative in the same paternal line as the missing person. The presence of the same Y-STRs in both samples tends to show that the remains are indeed those of the missing person.
221. Eugene A. Foster et al., Jefferson Fathered Slave’s Last Child, 396 Nature 27 (1998), https://doi.org/10.1038/23835. In response to letters emphasizing the inability of markers on the Y chromosome to distinguish between members of the Jefferson clan (including any illegitimate ones, white or black), the authors of the study noted that the genetic information was much more probable if the President rather than one of his sister’s children fathered Sally’s last child. The paper’s title, they acknowledged, was misleading. Eugene A. Foster et al., The Thomas Jefferson Paternity
Analytical Methods and Categorical Matches
Typically, the loci used in constructing a Y haplotype for forensic applications are STRs. Up to 30 are in use. For capillary electrophoresis, the same instruments and software used for routine STR testing are employed. Large peaks in an electropherogram that do not seem to be artifacts are interpreted as the alleles of the haplotype.222 As with autosomal STR profiling, the common practice for comparing a suspect’s profile to a trace-evidence one is the two-stage framework of a categorical match or no-match conclusion followed by a statistical evaluation. For instance, the Department of Justice’s Uniform Language for Testimony and Reports (ULTR) for Y-STRs states that an examiner “may” testify to an inclusion (meaning “included, or cannot be excluded”), exclusion, or inconclusive (because of an “indistinguishable mixture” or a possible “mutational event”).223 A declared match must be accompanied by some kind of “quantitative statement describing the weight of the evidence.”224
Case, 397 Nature 32 (1999), https://doi.org/10.1038/16177; see also Eliot Marshall, Which Jefferson Was the Father?, 283 Science 153 (1999), https://doi.org/10.1126/science.283.5399.153a.
222. Analysts need to be aware of certain quirks with Y-STR profiling to avoid some possible misintepretations of electropherograms. The International Society of Forensic Genetics explains that
[S]ome mutational effects rarely seen in autosomal STRs are more pronounced in Y-STRs. Especially large-scale deletions, insertions and conversions are responsible for higher numbers of Null . . . and multiple alleles at certain loci. Some markers included in commercial kits show always more than one allele because the sequence has identical copies on the Y chromosome (e.g. DYS385, DYF387S1). One or several Y-STR loci per haplotype can be involved in such mutation events. This is of forensic relevance, because a pattern can be erroneously interpreted as allelic drop-out, DNA contamination[,] and mixture, which may affect the evidential value of a DNA profile. Lutz Roewer et al., DNA Comm’n of the Int’l Soc’y of Forensic Genetics (ISFG): Recommendations on the Interpretation of Y-STR Results in Forensic Analysis, 48 Forensic Sci. Int’l: Genetics 102308, at 2 (2020), https://doi.org/10.1016/j.fsigen.2020.102308 (citations omitted). SWGDAM grandly declares that “[t]he laboratory should establish a method based on validation to document the designation of a peak as an artifact or an allele.” Scientific Working Group on DNA Analysis Methods, Interpretation Guidelines for Y-Chromosome STR Typing by Forensic DNA Laboratories § 4.1 (Mar. 2, 2022), https://perma.cc/ZZR4-FPND. It discusses the difficulties further in related and supplemental documents.
223. U.S. Dep’t of Just., Uniform Language for Testimony and Reports for Forensic Y-STR DNA Examinations 2–3 (Dec. 12, 2022) (https://perma.cc/LE6X-FS87) [hereinafter Y ULTR].
224. Id. at 3 (“An examiner shall provide a quantitative statement describing the weight of the evidence for all comparisons in which a known male is included as a possible contributor to the Y-STR typing results obtained from a probative evidentiary sample. This statement shall be provided regardless of the number of alleles detected or the magnitude of the resulting quantitative value.”). The ISFG is more lenient. Roewer et al., supra note 222, at 2 (“Basically, examiners are expected to prepare reports with the minimum requirement of a qualitative statement on the Y-STR test result.”). But see Y ULTR, supra note 223, at 5 (“When a suspect is identified that matches the Y-STR profile, we recommend the formulation of hypotheses according to the likelihood approach described by Evett and Weir.”); see also Ian W. Evett & Bruce S. Weir, Interpreting DNA Evidence: Statistical Genetics for Forensic Scientists (1998).
In court, laboratory performance issues and the manner in which inclusionary findings are presented are more likely to be raised than are any fundamental objections to the science and technology for determining whether two Y-STR profiles are the same. Reasoning that Y-STRs are just another set of markers involving the same principles and procedures as the previously accepted autosomal STRs, courts have upheld the admission of matching Y-STRs.225 Likewise, actions for postconviction Y-STR testing tend to be resolved on the assumption that this form of DNA testing is admissible.226
Population Genetics and Statistics
An exclusion reflects the analyst’s belief that differences between the alleles in the profiles are so extensive that they would never be seen in samples within the same paternal lineage. The inference that follows is clear: A correct exclusion means the suspect is not the source. However, the implication of an inclusion or match is not obvious without further information. If the inclusion is correct, the haplotypes are the same. But what follows from that? Does the shared haplotype occur once in a blue moon in the suspect population, or does it pop up all the time? Thus, an inclusion requires a second stage of statistical analysis to know the degree to which the finding, even if correct, tends to prove that the defendant is the source of the trace-evidence sample.227 The additional information
225. E.g., Shabazz v. State, 592 S.E.2d 876, 879 (Ga. Ct. App. 2004); Commonwealth v. Jacoby, 170 A.3d 1065, 1091–95 (Pa. 2017) (nothing novel about Y-STRs); Curtis v. State, 205 S.W.3d 656, 660–61 (Tex. Ct. App. 2006); Commonwealth v. Clark, 34 N.E.3d 1, 12 (Mass. 2015) (stating “that the results of DNA testing using the Y–STR method are admissible in Massachusetts courts” on the basis of a case with autosomal-STR testing).
226. E.g., United States v. MacDonald, 37 F. Supp. 3d 782 (E.D.N.C. 2014) (motion for Y-STR typing under the Innocence Protection Act of 2004, 18 U.S.C. § 3600, denied as untimely); Clark, 34 N.E.3d 1 (denial of a motion for Y-STR testing under a state statute was error).
227. An “inconclusive” means that the DNA test should not be used to decide whether the suspect is the source. It reflects the belief that even if the samples are in the same lineage, the observed differences are not extremely improbable considering the possibility of a mutation or other event. Neither are they very improbable if the samples come from different paternal lines. No inference follows. Yet, DNA analysts sometimes speak of the proportion of a population that would be excluded by haplotype testing as if it were simply 1 minus the proportion of fully matching haplotypes in a reference database for that population. E.g., State v. Polizzi, 924 So.2d 303, 308–09 (La. Ct. App. 2006) (The DNA expert “concluded that the two profiles were consistent with each other, meaning that the Defendant or any of his paternal relatives could not be excluded as having been a donor to the sample from the victim. . . . 99.7 percent of the Caucasian population, 99.8 percent of the African American population, and 99.3 percent of the Hispanic population could be excluded as donors of the DNA in the sample.”). But if haplotypes with slightly different STRs—they match except for one mutation—are considered inconclusive, men with those haplotypes cannot be excluded. Hence, the one-off haplotypes should be added in computing the fraction of the population that “could not be excluded.” Whether such slightly different haplotypes
needed to evaluate a match comes from population genetics and statistics. To provide this information, researchers have gathered DNA samples from populations both within the United States and across the world and examined them for Y-STRs and Y-SNPs.228 The goal is to draw on these “reference databases” to estimate the unrelated-man-haplotype frequency or match probability.
Haplotype frequency or probability estimates
Because the entire haplotype (for example, a set of perhaps a dozen to 30 STRs strung out across the Y chromosome) is inherited as a package, it is neither necessary nor appropriate to combine any allele frequencies as is done with autosomal STRs. The haplotype itself is like a single allele, and one can simply count how often it occurs in a sample of unrelated men.229 As discussed in the Reference Guide on Statistics and Research Methods, in this manual, dividing the count (x) by the sample size (n) gives the sample proportion (p = x/n). For instance, in State v. Jones,230 a Y-STR haplotype was present in DNA from fingernail clippings from a woman who had been murdered in the Salt Lake City area and who evidently had struggled with her attacker. A laboratory director testified to zero occurrences of the haplotype in a database of size n = 8,028.231 The sample proportion in Jones was therefore p = 0/8,028 = 0.
Two variations on the sample proportion x/n as an estimator of a population frequency have some popularity in the forensic-genetics community. Following the testing in the case, the number of times that the haplotype has been seen is no longer x out of n. Now it is x + 1 out of the n + 1 men with known haplotypes. In Jones, this number is 1/8,029, or about 0.012%. Internationally, this “augmented” statistic is typically used as an estimator of the population frequency.232 There also are arguments for using (x + 2)/(n + 2) or (x + 2)/(n + 3), which would be about 0.025% in Jones.233
were treated as “consistent with” one another in the percentages recited in Polizzi is ambiguous at best.
228. The freely available Y Chromosome Haplotype Reference Database contains data from “more than 1,300 local population samples with more than 270,000 haplotypes in 136 countries.” Lutz Roewer, Using the YHRD Database for Casework Analysis, ISHI Rep. Apr. 2019, https://perma.cc/25AP-F6VJ.
229. In this context, “unrelated people” means individuals with a different paternal lineage.
230. 345 P.3d 1195 (Utah 2015).
231. Id. at 1208 n.42.
232. Roewer et al., supra note 222, at 3.
233. See Michael Coble et al., Non-autosomal Forensic Markers, in Forensic DNA Evidence Interpretation 315, 331 (John Buckleton et al. eds., 2d ed. 2016). The SWGDAM Y chromosome guidelines only state that “[t]he laboratory should determine which of the following methods will be used.” SWGDAM, supra note 222, § 9.2.2.
These sample proportions do not account for sampling error—the inevitable differences between random samples and the population from which they are drawn. Finding no matches in a sample of 8,028 men hardly means that there would be no other paternal line with a matching haplotype among the hundreds of thousands of men in the Salt Lake City area. If there are any, a different sample might have produced some of them. One solution to this “zero numerator” problem234 (and to the problem of quantifying sampling error generally) is to supply a confidence interval for plausible values of the population frequency. Testimony that incorporates this concept is common. The laboratory director in Jones testified “that ‘approximately 99.6 percent of . . . the male population can be excluded’ as a contributor of the DNA sample but that Mr. Jones could not be excluded” and that “read another way, the frequency of Mr. Jones’s DNA profile ‘is equivalent to one in 2681 individuals.’ He explained this means that ‘every time you test . . . a male [in a different paternal line], the probability of that person having that particular DNA profile is approximately one in 2681.’”235
How did the witness get from about 0/8,000 to 1/2,681? The latter figure is an answer to the following question: How large would the population proportion have to be before we would expect to encounter random samples of size 8,028 that are devoid of the haplotype (as in our one database of this size) at least 5% of the time? If the proportion were much larger than 1/2,681, then random samples with no matching haplotype would be rarer. As such, an estimate of 1/2,681 for the population haplotype frequency is not plainly incompatible with the finding that the haplotype is not in the database. It is the upper end of a 95% “confidence interval.”236
Case law generally accepts the confidence-interval procedure for estimating the frequency of a Y haplotype in reference-population databases.237 It also rejects arguments that quantitative analyses are unfairly prejudicial; however,
234. Kaye & Stern, supra note 12, section titled “Other Situations.”
235. State v. Jones, 345 P.3d 1195, 1208 (Utah 2015) (omissions in original).
236. When drawing from a population whose proportion is 1/2,681, the probability of a simple random sample of size 8,028 with no matching haplotypes is 5%. For a population proportion of 1/1,744, the probability of no haplotypes is 1%, so it can be called the upper limit of a 99% confidence interval. A 99% interval may sound better than a 95% interval, but the larger proportion of 1/1,744 is less compatible with the database. It generates random samples that lack the haplotype less often.
The calculations here use the Poisson approximation. Other ways to produce confidence intervals are sensible. Coble et al., supra note 233, at 338–39; Kaye & Stern, supra note 12, section titled “Other Situations.” The SWGDAM guidelines cite one procedure. SWGDAM, supra note 222, § 9.2.3.
237. E.g., Commonwealth v. Jacoby, 170 A.3d 1065 (Pa. 2017); State v. Tucker, 920 N.W.2d 680 (Neb. 2018); State v. Jones, 345 P.3d 1195 (Utah 2015); State v. Bander, 208 P.3d 1242, 1255 (Wash. Ct. App. 2009) (noting that defendant made no showing of a scientific controversy); cf. Commonwealth v. Lally, 46 N.E.3d 41, 52–53 (Mass. 2016) (confidence interval not required if point estimate is explained clearly).
some opinions admonish trial judges and counsel to avoid exaggerating the power of Y haplotypes for source attribution.238 The scientific consensus seems to be that the “counting method” and its confidence intervals are overly conservative for rare haplotypes.239 As such, further refinements have been developed,240 and population-genetics models that take into account the way haplotypes evolve in reproducing populations may be the most technically defensible basis for estimating frequencies and likelihood ratios.241
Likelihood ratios (LRs)
Likelihood ratios (defined in the section titled “Mixtures” later in this reference guide) are recommended for Y haplotypes by the International Society of Forensic Genetics (ISFG),242 are recognized by SWGDAM,243 and are advocated for reporting DNA and other forensic-science findings by many scholars and other groups.244 LRs have the potential advantage of clarifying the weight of the evidence with respect to a range of alternative hypotheses. For example, the contribution of the analysis of the Y haplotypes to the historical debate over the paternity of Sally Hemings’s children (see section titled “Forensic Value” above)
238. Jones, 345 P.3d at 1249.
239. Roewer et al., supra note 222, at 2–3; Coble et al., supra note 267, at 330–35.
240. One such adjustment is the “kappa method.” Butler, supra note 113, at 423–24; Coble et al., supra note 267, at 332; Roewer et al., supra note 222, at 3.
241. The discrete Laplace method has been said to be the most robust. Coble et al., supra note 267, at 333; see also Roewer et al., supra note 222, at 3 (“established in practice” internationally). Another consideration is the adjustment for population structure when the reference database comes from an amalgam of preferentially reproducing subgroups in a larger population (see supra section titled “Could an Unrelated Person Be the Source?”). For advice, see Coble et al., supra note 233, at 329, 330, 334–35; Roewer et al., supra note 222, at 5.
242. Roewer et al., supra note 222, at 5. The ISFG gives a generic example of possible wording to explain the number:
The Y-STR profile detected in the crime stain is LR times more probable to observe under hypothesis H1 than under hypothesis H2. This notwithstanding, paternal relatives have a high probability to have the same Y-STR profile and will in that case have the same likelihood ratio (LR). Id.
243. SWGDAM, supra note 222, § 9.2.5 (“The laboratory should determine if likelihood ratios will be used to provide quantitative assessments of the value of the matches using relevant populations.”).
244. See section titled “Frequencies, Probabilities, and Prejudice” above. The Department of Justice ULTR demands that “[a]n examiner shall provide a quantitative statement describing the weight of the evidence for all comparisons in which a known male is included as a possible contributor to the Y-STR typing results obtained from a probative evidentiary sample.” Y ULTR, supra note 223, at 3. Presumably, the LR can be the “quantitative statement [of] weight” for inclusions, but the ULTR does not address its use for exclusions (where the LR is very large for the hypothesis that a man other than the defendant is the source as opposed to the hypotheses that the defendant is the source) and inconclusives (where the LR is 1).
might have been clearer from the outset had the researchers made statements for each hypothesis about paternity—for example:
- The Jefferson haplotype is at least 100 times more probable if the president was the father of Eston Hemings Jefferson than if one of the president’s sister’s boys was the father;
- The Jefferson haplotype has the same probability if the president was the father as it does if the president’s brother was the father.
Similarly, in State v. Jones,245 the likelihood approach to reporting results would transform the testimony that “this means that 99.92 percent of the male population could be excluded as a possible donor” into:
- The haplotype is about 2,680 times more probable if Mr. Jones is its source than if any particular man not in Mr. Jones’s paternal family tree is the source; and
- The haplotype has the same probability if Mr. Jones is its source as it does if any other man in his paternal family tree (a father, child, uncle, certain cousins, nephews, etc.) is the source.
In addition to its clarity with regard to hypotheses in the case, the broader likelihood ratio framework does not require the match/no-match categories and rules for “inconclusives.” It can better describe the evidentiary value of haplotypes that are one mutation away from a clear match246 and can be applied to ambiguous data from low-template DNA.247 However, questions can remain as to the computation of likelihood ratios in these more challenging cases.
Mitochondrial DNA
Mitochondria and Their Genomes
Mitochondria are small structures, with their own membranes, found inside the cell but outside its nucleus. Within these organelles, molecules are broken down to supply energy. Mitochondria have a small genome—a circle of 16,569 nucleotide base pairs that includes only 37 genes. They started out as bacteria that were engulfed by cells billions of years ago. Many of their original genes migrated to
245. See supra section titled “Genetic Principles.”
246. Coble et al., supra note 233, at 340; cf. supra note 227 (on the impact of “inconclusives” on exclusion probabilities).
247. Coble et al., supra note 233, at 342–44.
the nuclear chromosomes,248 where they produce proteins and RNAs for the mitochondria.249 However, the sequences currently found in the mitochondria themselves bear little relation to the comparatively monstrous chromosomal genome in the cell nucleus. In particular, they are not physically or statistically associated with the forensic STRs.
Forensic Value
Mitochondrial DNA (mtDNA) has four features that make it useful for forensic DNA testing. First, the typical cell, which has but one nucleus, contains hundreds or thousands of nearly identical mitochondria. Hence, for every copy of chromosomal DNA, there are hundreds or thousands of copies of mitochondrial DNA. This makes it possible to detect mtDNA in samples, such as bone and hair shafts, that contain too little nuclear DNA for conventional typing.
Second, two hypervariable regions that tend to be different in different individuals lie within the control region or D-loop (displacement loop) of the mitochondrial genome.250 These regions extend for a bit more than 300 base pairs each—short enough to be typable even in highly degraded samples such as very old human remains.
Third, mtDNA comes solely from the egg cell.251 For this reason, mtDNA is inherited maternally, with no paternal contribution:252 Full siblings, maternal half-siblings, and others related through maternal lineage normally possess the same mtDNA sequence. This feature makes mtDNA particularly useful for
248. James B. Stewart & Patrick F. Chinnery, Extreme Heterogeneity of Human Mitochondrial DNA from Organelles to Populations, 22 Nature Rev. Genetics 106, 106–07 (2021), https://doi.org/10.1038/s41576-020-00284-x.
249. Whole-genome sequencing of parents and their children establishes that other segments of mitochondrial DNA (mtDNA) continue to find their way into the nuclear genome, creating new nuclear mtDNA segments (NUMTs) in some cells. E.g., Wei Wei et al., Nuclear-embedded Mitochondrial DNA Sequences in 66,083 Human Genomes, 611 Nature 105 (2022), https://doi.org/10.1038/s41586-022-05288-7.
250. A third, somewhat less polymorphic region in the D-loop, can be used for additional discrimination. The remainder of the control region, although noncoding, consists of DNA sequences that are involved in the transcription of the mitochondrial genes. These control sequences are essentially the same in everyone (monomorphic).
251. The relatively few mitochondria in the spermatozoan that fertilizes the egg cell soon degrade and are not replicated in the multiplying cells of the pre-embryo.
252. The possibility of paternal contributions to mtDNA in humans is discussed in Alistair T. Pagnamenta et al., Biparental Inheritance of Mitochondrial DNA Revisited, 22 Nature Rev. Genetics 477 (2021), https://doi.org/10.1038/s41576-021-00380-6 (“Evidence for a biparental mode of . . . inheritance has been sparse and remains controversial. Recent studies using a range of complementary techniques do not support paternal transmission of mtDNA and highlight the co-amplification of rare, concatenated nuclear mtDNA segments as a technical artefact that may explain previous observations.”).
associating persons related through their maternal lineage. It has been exploited to identify the remains of the last Russian tsar and other members of the royal family, of soldiers missing in action, and of victims of mass disasters.253
Finally, point mutations accumulate, particularly in the noncoding D-loop, without altering how the mitochondrion functions. Hence, a single individual can develop distinct internal populations of mitochondria. Single nucleotide and other variants can occur within a cell, between cells in a given tissue, and between cells from different tissues and organs.254 Such “heteroplasmy” has been observed in healthy humans, with an average of one heteroplasmic site per individual.255 As discussed below, heteroplasmy complicates the interpretation of differences in mtDNA sequences.256 Yet, it is mutations that make mtDNA polymorphic and hence useful in identifying individuals. Over time, mutations in egg cells can propagate to later generations, producing more heterogeneity in the inherited mitochondrial genomes in the human population.257 This polymorphism allows scientists to compare mtDNA from crime scenes to mtDNA from given individuals to ascertain whether the tested individuals are within the maternal line (or another coincidentally matching maternal line) of people who could have been the source of the trace evidence.
Analytical Methods and Categorical Matches
The small mitochondrial genome can be analyzed with sequencing methods or with microarrays that give the order of some or all the base pairs.258 The traditional Sanger-sequencing technology usually was done on just the hypervariable parts of the coding region. Cheaper and faster massively parallel sequencing is replacing that procedure, increasing the ability of laboratories to detect heteroplasmy and better distinguish among different maternal lineages in the population.259 The laboratory first generates descriptions of the sequences of two
253. See, e.g., Silent Witness: Forensic DNA Analysis in Criminal Investigations and Humanitarian Disasters (Henry Ehrlich et al. eds., 2020).
254. “Length heteroplasmy” (most often in the form of simple repeats such as CC . . .) is common but harder to characterize precisely in sequencing studies.
255. Alison Barrett et al., Pronounced Somatic Bottleneck in Mitochondrial DNA of Human Hair, 375 Phil. Trans. Roy. Soc’y B 20190175, at 2 (2019), https://doi.org/10.1098/rstb.2019.0175.
256. See infra section titled “Population Genetics and Statistics.”
257. Estimates of the average mutation rate range up to around 1 per 70 generations. Mikkel M. Andersen & David J. Balding, How Many Individuals Share a Mitochondrial Genome?, 14 PLoS Genetics e1007774 (2018), https://doi.org/10.1371/journal.pgen.1007774.
258. See supra sections titled “Sequence-Specific Probes and Microarrays” and “Sequencing and SNPs.”
259. For a survey of the major methods, see Bethany Forsythe et al., Methods for the Analysis of Mitochondrial DNA, 3 WIREs Forensic Sci. e1388 (2021), https://doi.org/10.1002/wfs2.1388.
samples—say, DNA extracted from a hair shaft found at a crime scene and hairs plucked from a suspect.260 Let’s assume that there is no issue as to whether the technology has been properly applied with suitable controls,261 and the factfinder can be confident that the sequences as reported truly represent the order of the base pairs in the mtDNA of the hairs. What should the factfinder make of the two sequences?
Most analysts use match/no-match categories to describe the result of a comparison.262 The Department of Justice currently favors the words “[c]annot be excluded (i.e., inclusion, or included) [and] [e]xclusion (i.e., excluded).”263 As will be explained in the section below titled “Heteroplasmy,” an intermediate category of “inconclusive” covers less-than-perfectly-matching sequences.264 In the simplest cases, the two sequences show a good number of differences (a clear exclusion), or they are identical (an inclusion). Suppose there is an exclusion. After explaining why a clear “exclusion” means that such a large difference almost certainly cannot occur for two hairs from people within the same lineage, an analyst’s testimony on direct examination could end. But if the analyst were to stop at the same point when declaring an inclusion, the factfinder would be left with no scientific information on how common or rare such matching sequences are for different lineages in the relevant population. Such inclusion-only testimony invites the objection that the congruent sequences are of unknown probative value and might be given more weight than they deserve.265 To more
260. Mitotyping often can exclude individuals as the source of stray hairs even when the hairs are microscopically indistinguishable. Nonetheless, gross or microscopic hair comparison can be useful, as a screening test, to exclude a suspect without the need for DNA analysis. See ASTM E3316–22, Standard Guide for Forensic Examination of Hair by Microscopy (2022).
261. See Walther Parson et al., DNA Comm’n of the Int’l Soc’y for Forensic Genetics: Revised and Extended Guidelines for Mitochondrial DNA Typing, 13 Forensic Sci. Int’l: Genetics 134, 135–36 (2014), https://doi.org/10.1016/j.fsigen.2014.07.010; Scientific Working Group on DNA Analysis Methods, SWGDAM Interpretation Guidelines for Mitochondrial DNA Analysis by Forensic DNA Testing Laboratories § 1 (Apr. 23, 2019) [hereinafter SWGDAM MtDNA Guidelines].
262. E.g., Lewis v. State, 889 So.2d 623, 673 (Ala. Ct. Crim. App. 2003) (FBI expert testified that “[t]he fourth step is to compare the order of the bases, i.e., the sequence, to other samples to determine if there is a ‘match.’”); see also SWGDAM MtDNA Guidelines, supra note 261, § 3.1.
263. U.S. Dep’t of Just., Uniform Language for Testimony and Reports for Forensic Mitochondrial DNA Examinations 2 (Dec. 12, 2022), https://perma.cc/J6V4-Z4EG [hereinafter MtDNA ULTR]. This terminology document does not single out “match” or words based on that stem as impermissible. It defines these labels as “an examiner’s conclusion” with no specification for how the examiner should reach these conclusions.
264. In principle, a likelihood ratio is better suited to cases in which there are slight sequence differences (and could be used in all situations). See Coble et al., supra note 233, at 319, 324–41; cf. supra section titled “Likelihood ratios (LRs)” (on likelihood ratios for Y-STRs).
265. But see Commonwealth v. Chmiel, 30 A.3d 1111, 1158 (Pa. 2011) (suggesting that “statistical analysis is . . . unnecessary”).
adequately inform the court or jury of the implications of a match, forensic scientists invoke principles of population genetics and statistics.266
Population Genetics and Statistics for Mitochondrial DNA
Population databases
As with Y-STRs, to indicate the significance of the match, analysts usually estimate the frequency of the sequence in some population. It is not necessary to combine any allele frequencies because the entire mtDNA sequence, whatever its internal structure may be, is inherited as a single unit (a haplotype). In other words, the sequence itself is like a single allele, and one can simply count how often it occurs in a sample of unrelated people.267
Laboratories therefore refer to databases of mtDNA sequences to see how often the type in question has been seen before and to compute the probability of a matching haplotype in a random draw from the population given that it has been observed in the case under investigation.268 The issues are essentially the same as those for Y haplotypes.269 Key questions are which reference population or populations to use;270 what estimator to use for the sequence frequency or match probability;271 how to account for sampling error in this estimate;272
266. The Department of Justice expects analysts in its laboratories to “provide a quantitative statement describing the weight of the evidence for all inclusions regardless of the magnitude of the resulting quantitative value.” MtDNA ULTR, supra note 263, at 3.
267. In this context, “unrelated people” means individuals with a different maternal lineage.
268. The publicly available European DNA Profiling Group (EDNAP) mtDNA Population Database (EMPOP), which houses quality-controlled sequence data from populations across the world, was established in 2006. Walther Parson & Arne Dür, EMPOP—A Forensic mtDNA Database, 1 Forensic Sci. Int’l: Genetics 88 (2007), https://doi.org/10.1016/j.fsigen.2007.01.018.
269. See supra section titled “Population Genetics and Statistics.”
270. See Parson et al., supra note 261, at 140 (“Local, regional and continental databases may all be searched and reported to guide evaluation of the evidence. Additionally, frequencies from geographic databases of relevant differentiated populations may be reported (as in the United States where reports often list separately search results from major population groups).”); supra section titled “Haplotype frequency or probability estimates.”
271. See Parson et al., supra note 261, at 140; supra section titled “Haplotype frequency or probability estimates” (noting most of the alternatives). In State v. Pappas, 776 A.2d 1091 (Conn. 2001), the defendant maintained that the usual sample proportion is inadmissible because an estimate that tosses in the haplotype in the case itself is more appropriate. The Connecticut Supreme Court responded that “using zero as the numerator rather than one . . . goes to the weight of the evidence and not to its admissibility.” Id. at 1109.
272. See Parson et al., supra note 261, at 140 (“approaches that empirically take into account the haplotype distribution of the database in question (e.g. the Kappa model . . .) may best represent the strength of the mtDNA evidence”); SWGDAM MtDNA Guidelines, supra note 261, § 4.2.4 (binomial distribution); supra section titled “Haplotype frequency or probability estimates”; infra note 275.
whether and how to adjust for population structure;273 whether to present likelihood ratios, including one that combines the information in mitochondrial haplotypes with that from Y haplotypes (or both Y and autosomal haplotypes);274 and how to present or explain the statistical assessment to a factfinder.275
Heteroplasmy
The simple inclusion–exclusion approach must be modified to account for the fact that the same individual can have detectably different (albeit very similar) mitotypes in different tissues or even in different cells in the same tissue. To understand the implications of heteroplasmy, we need to understand how it comes into existence. Heteroplasmy can occur because of mtDNA mutations during the division of adult cells, such as those at the roots of hair shafts. These new mitotypes are confined to the individual. They will not be passed on to future generations. Heteroplasmy also can result from a mutation contained in the egg cell that grew into an individual. Such mutations can make their way into succeeding generations, establishing new mitotypes in the population. But this is an uncertain process. Egg cells contain many mitochondria, and the
273. See supra section titled “Haplotype frequency or probability estimates.” Compare Coble et al., supra note 233, at 334–35 (describing a procedure) with SWGDAM MtDNA Guidelines, supra note 261, § 4.3 (“However, determination of an appropriate theta (θ) value is complicated by the variety of primer sets, covering different portions of HV1 and/or HV2, which may be applied to forensic casework. SWGDAM has not yet reached consensus on the appropriate statistical approach to estimating θ for mtDNA comparisons.”).
274. See Parson et al., supra note 261, at 140; SWGDAM MtDNA Guidelines, supra note 261, §§ 4.4 & 4.5.
275. Apparently, SWGDAM would dispense with estimates of sampling error. SWGDAM MtDNA Guidelines, supra note 261, § 4.2.3 (“Reporting an mtDNA haplotype sample frequency without a confidence interval is acceptable as a factual statement regarding observations in the database.”). But the conclusion that judges or jurors can make suitable use of a sample proportion without an assessment of statistical uncertainty is not a strictly scientific judgment. A court might need to consider whether it is sufficient to leave it to the jury to decide how to weigh the fact of the match and the absence of the same sequence in a convenience sample that might—or might not—be representative of the local population.
As with Y haplotyping (supra note 227), the use of exclusion probabilities that do not account for inconclusives could be misleading. In Vaughn v. State, 646 S.E.2d 212, 215 (Ga. 2007), for instance, the statement that a suspect “cannot be excluded” when “there is a single base pair difference” was transformed into “a match.” These inconclusive sequences contribute to the number of people who would not be excluded. Therefore, in Pappas, it was misleading to conclude “that approximately 99.75% of the Caucasian population could be excluded as the source of the mtDNA in the sample.” 776A.2d 1091, 1104 (Conn. 2001) (footnote omitted). This percentage neglects the individuals whose mtDNA sequences are off by one base pair. Along with the 0.25% who are included because their mtDNA matches completely, these one-off people would not be excluded. Of course, the difference may be fairly small. In Pappas, a defense expert reported that the actual nonexclusion rate was still “99.3 percent of the Caucasian population.” Id. at 1105 (footnote omitted).
mature egg cell will not contain just the mutation—it will house a mixed population of the old-style mitochondria and a number of the mutated ones (with DNA that usually differs from the original at a single base pair). Figuratively speaking, the original mtDNA sequence and the mutated version fight it out for several generations until one of them becomes “fixed” in the population. In the interim, the progeny of the mutated egg cell will harbor both strains of mitochondria.
When mtDNA from a single-source crime-scene sample is compared to a suspect’s sample, there are three possibilities: (1) neither sample is detectably heteroplasmic; (2) one sample displays heteroplasmy, but the other does not; (3) both samples display heteroplasmy. In each scenario, the comparison can produce an exclusion or an inclusion:
- Neither sample heteroplasmic. In the first situation, if the sequence in the crime-scene sample is markedly different from the sequence in the suspect’s sample, then the suspect is excluded. But heteroplasmy could be the reason for a difference of only a single base or so. For example, the sequence in a hair shaft coming from the suspect could be a slight mutation of the dominant sequence in the suspect. Therefore, the FBI treats a difference at a single base pair as inconclusive.276 When the one mtDNA sequence characteristic of each sample is identical, the issue becomes how to use the reference database of mtDNA sequences, as discussed above.
- Suspect’s sample heteroplasmic, crime-scene sample not. One version of the second scenario arises when heteroplasmy is seen in the suspect’s tissues but not in the crime-scene sample. If the crime-scene sequence is not close to either of the suspect’s sequences, then the suspect is excluded. If it is identical to one of the suspect’s sequences, then the suspect is included, and a suitable reference database should indicate how infrequent such an inclusion would be. If crime-scene DNA is one base pair removed from either of the suspect’s sequences, then the result is inconclusive.
- Both samples heteroplasmic. In this third scenario, multiple sequences are seen in each sample. To keep track of things, we can call the sequences in the crime-scene sample C1 and C2, and those in the suspect’s sample S1 and S2. If either C1 or C2 is very different from both S1 and S2,
276. SWGDAM MtDNA Guidelines, supra note 261, § 3.1. Where did the one-base-pair rule come from? With traditional Sanger sequencing of parts of the control region of the mitogenome, heteroplasmy at a single point seems to occur, on average, in approximately 6% of the control-region sequences from around the world; heteroplasmy at multiple sites is seen in less than 1% of sequences. Jodi A. Irwin et al., Investigation of Heteroplasmy in the Human Mitochondrial DNA Control Region: A Synthesis of Observations from More Than 5000 Global Population Samples, 68 J. Molecular Evolution 516 (2009), https://doi.org/10.1007/s00239-009-9227-4.
- the suspect is excluded. If C1 and C2 are the same as S1 and S2, the suspect is included. Because detectable heteroplasmy is not very common, this inclusion is stronger evidence of identity than the simple match in the first scenario. Finally, in some of the situations in which C1 or C2 are merely very close to S1 or S2, the comparison will be inconclusive.
Emerging Questions for Courts
A number of courts have rejected objections that Sanger sequencing of hypervariable regions of the mitochondrial DNA genome does not comport with Frye277 or Daubert.278 But forensic-DNA laboratories are adopting some of the massively parallel sequencing (MPS) methods, described in the section above titled “Sequencing methods,” that can work with smaller quantities of mtDNA and that can efficiently sequence the entire mitogenome.279 The widespread use of MPS methods in clinical medicine and genetic research indicates scientific acceptance and validity at a general level (see “Sequencing methods”), but courts still may encounter questions about the rate of errors in the “reads,” what steps are taken to correct them, precautions against misinterpreting nuclear mtDNA segments (NUMTs) as present in mitochondria, and more. Much depends on the equipment and its operation.280 Published validity studies from biotechnology companies developing the sequencers and kits, from the individual forensic
277. E.g., Magaletti v. State, 847 So. 2d 523, 528 (Fla. Dist. Ct. App. 2003) (“[T]he mtDNA analysis conducted [on hair] determined an exclusionary rate of 99.93 percent. In other words, the results indicate that 99.93 percent of people randomly selected would not match the unknown hair sample found in the victim’s bindings.”); People v. Sutherland, 860 N.E.2d 178, 271–72 (Ill. 2006); People v. Holtzer, 660 N.W.2d 405, 411 (Mich. Ct. App. 2003); Wagner v. State, 864 A.2d 1037, 1043–49 (Md. Ct. Spec. App. 2005) (mtDNA sequencing admissible despite contamination and heteroplasmy).
278. E.g., United States v. Beverly, 369 F.3d 516, 531 (6th Cir. 2004) (“The scientific basis for the use of such DNA is well established.”); United States v. Coleman, 202 F. Supp. 2d 962, 967 (E.D. Mo. 2002) (“‘[a]t the most,’ seven out of 10,000 people would be expected to have that exact sequence of As, Ts, Cs, and Gs”), aff’d, 349 F.3d 1077 (8th Cir. 2003); Pappas, 776 A.2d at 1095; State v. Underwood, 518 S.E.2d 231, 240 (N.C. Ct. App. 1999); State v. Council, 515 S.E.2d 508, 518 (S.C. 1999); State v. Griffin, 384 P.3d 186, 202 (Utah 2016).
279. The methods being implemented are not the most advanced ones that have been proposed for mitochondrial research. It is technically possible to zoom in on the mitogenome (as a single molecule, without PCR amplification) and sequence it in a single “read.” Ieva Keraite et al., A Method for Multiplexed Full-Length Single-Molecule Sequencing of the Human Mitochondrial Genome, 13 Nature Communications 5902 (2022), https://doi.org/10.1038/s41467-022-33530-3 (using CRISPR-Cas9 and claiming to overcome the relatively large rate of read errors in single-molecule sequencing systems).
280. See, e.g., Yun Heo et al., Comprehensive Evaluation of Error-Correction Methodologies for Genome Sequencing Data, in Bioinformatics (Nakaya Helder ed., 2021); Nicholas Stoler & Anton
laboratories deploying them, and from academic researchers can be scrutinized.281 At least in early admissibility hearings, courts may wish to consider appointing independent expert advisers for this purpose.
Questions of the accuracy of the sequences and their interpretation also might emerge with respect to the “reliably applied” prong of the requirements for scientific evidence under Rule 702. The presence of and adherence to rigorous quality control and assurance systems might be significant.282 Moreover, all these matters are potential topics that can arise for jurors (and judges when they act as triers of fact). The practice followed in a case at bar can be compared to both minimally acceptable and recommended practices as articulated in the scientific literature and guidelines from expert groups.283
Courts have consistently held that neither the phenomenon of heteroplasmy nor the limitations in the statistical analysis preclude the forensic use of sequencing results under either Rule 702 or Rule 403.284 However, mitogenome MPS picks up more heteroplasmic sites than Sanger sequencing, and more individuals exhibit not just one, but several points of heteroplasmy.285 As a result, restricting the “inconclusive” category to only a single sequence-difference across the mitogenome will not prevent as many false exclusions. Despite the allure of purportedly definitive “exclusions” and “inclusions,” the gray zone of “inconclusive” sequence differences is increasingly hard to define with a simple rule of thumb for the entire mitogenome. Courts may need to inquire into what the literature demonstrates about the accuracy of the laboratory’s rule or procedure for assessing the extent to which sequence similarities and differences support conclusions (including judgments of “inconclusive”) about the maternal relatedness of the unknown individual whose mtDNA was recovered and the suspect’s comparison sample.
Nekrutenko, Sequencing Error Profiles of Illumina Sequencing Instruments, 3 Nucleic Acids Rsch. Genomics & Bioinformatics lqab019 (2021), https://doi.org/10.1093/nargab/lqab019.
281. E.g., Michael D. Brandhagen et al., Validation of NGS for Mitochondrial DNA Casework at the FBI Laboratory, 44 Forensic Sci. Int’l: Genetics 102151 (2020), https://doi.org/10.1016/j.fsigen.2019.102151 (sequencing the control region); Jennifer Churchill Cihlar et al., Developmental Validation of a MPS Workflow with a PCR-Based Short Amplicon Whole Mitochondrial Genome Panel, 11 Genes 1345 (2020), https://doi.org/10.3390/genes11111345 (whole-mitogenome sequencing).
282. See supra section titled “Sample Collection and Laboratory Performance.”
283. See supra note 261.
284. E.g., Beverly, 369 F.3d at 531 (“[T]he mathematical basis for the evidentiary power of the mtDNA evidence was carefully explained, and was not more prejudicial than probative.”); Pappas, 776 A.2d 1091; Griffin, 384 P.3d at 202–03.
285. Jennifer A. McElhoe et al., Exploring Statistical Weight Estimates for Mitochondrial DNA Matches Involving Heteroplasmy, 136 Int’l J. Legal Med. 671, 695–96 (2022), https://doi.org/10.1007/s00414-022-02774-5.
Mixtures
What Are DNA Mixtures?
Samples of biological trace evidence recovered from crime scenes often contain a mixture of fluids or tissues from different individuals. Examples include vaginal swabs collected as sexual assault evidence, bloodstain evidence from scenes where several individuals shed blood, and objects that several people touched. However, not all mixed samples produce mixed STR profiles. Consider a sample in which 99% of the DNA comes from the defendant and 1% comes from a different individual. Even if some of the molecules from the minor contributor come in contact with a primer and the polymerase and an STR is amplified, the resulting signal might be too small to detect—the peak in an electropherogram will blend into the background. Because the vast bulk of the amplified STRs will come from the defendant’s DNA, the electropherogram may show only one STR profile. In these situations, the interpretation of the single DNA profile is the same as when 100% of the DNA molecules in the sample are the defendant’s.
When the mixtures are more evenly balanced among contributors, however, the STRs from multiple contributors can appear as “extra” peaks. As a rule, because DNA from a single individual can have no more than two alleles at each locus,286 the presence of three or more peaks at several loci indicates a mixture of DNA in the sample.287 Figure 6 shows another electropherogram from DNA recovered in People v. Pizarro.288 The fact that there are as many as four alleles at
286. This follows from the fact that individuals inherit chromosomes in pairs, one from each parent. See supra section titled “Sexual Reproduction and the Genome.” An individual who inherits the same allele from each parent (a homozygote) can contribute only that one allele to a sample, and an individual who inherits a different allele from each parent (a heterozygote) will contribute those two alleles. Finding three or more alleles at several loci therefore indicates a mixture.
287. On rare occasions, an individual exhibits a phenotype with three alleles at a locus. This can be the result of a chromosome anomaly (such as a duplicated gene on one chromosome or a mutation). A sample from such an individual is usually easily distinguished from a mixed sample. The three-allele variant is seen at only the affected locus, whereas with mixtures, more than two alleles typically are evident at several loci.
Individuals who are genetic chimeras also can produce DNA samples that could be mistaken for that of two different people. Chimerism refers to more than one cell line resulting from different zygotes (fertilized eggs). Chimerism can arise artificially, as a result of a blood transfusion or transplantation, or spontaneously, during the formation and development of an embryo. Chimerism: A Clinical Guide 3–48 (Nicole L. Draper ed., 2018). The implications for forensic-DNA identification are discussed in, e.g., David H. Kaye, Chimeric Criminals, 14 Minn. J.L. Sci. & Tech. 1 (2012); Erin Murphy, Inside the Cell: The Dark Side of Forensic DNA 246–49 (2015); Kayla Sheets & Robert Wenk, Relationship Testing and Forensics, in Chimerism: A Clinical Guide, supra, at 51–63.
 Source: Steven Myers and Jeanette Wallin, California Department of Justice, provided the image.
Source: Steven Myers and Jeanette Wallin, California Department of Justice, provided the image.That some loci in Figure 6, such as vWA, exhibit only two peaks does not preclude the existence of a two-person mixture. Both the victim and the rapist (on the state’s theory of the case) might share the same alleles at that locus, and the amplified product of the victim’s DNA might be masking that from the rapist. Both might be homozygous at that locus (each producing one peak). If it is credible that one or both alleles of the rapist’s vWA locus dropped out of the electropherogram (not likely with substantial quantities of DNA), still more scenarios consistent with the state’s theory of the case could explain why there are only two peaks at the vWA locus. Enumerating and assessing such possibilities leads to the topic of “mixture deconvolution.”
some loci and that many of the peaks match the victim’s suggests that the sample is a mixture of the victim’s and another person’s DNA. Furthermore, the presence of two peaks at the amelogenin locus (labeled AMEL in the electropherogram) shows that male DNA is part of the mixture. Because all the peaks that do not match the victim are part of the defendant’s STR profile, the mixture is consistent with the state’s theory that the defendant raped the victim.
Have Strategies for Simplifying Mixtures Interpretation Been Pursued?
A pioneer in the development of DNA typing methods and their interpretation had this advice for forensic laboratories: “Don’t do mixture interpretations unless you have to.”289 If a laboratory has other samples that do not show evidence of mixing, it may be able to avoid fully deciphering mixed profiles. Even across a single stain, the proportions of a mixture can vary, and it might be possible to extract a DNA sample that does not produce a mixed profile.
In sexual assault cases, it sometimes is possible to separate sperm from the male and female epithelial cells on vaginal, oral, or rectal swabs and then analyze the sperm DNA by itself.290 When this procedure works well, and only one man’s sperm are present, the electropherogram should display one clear profile with little or no interference from the female DNA. And even with sperm from more than one man, the analysis might be simpler because the victim’s DNA no longer can mask alleles from the sperm. Also, in sexual assault (and other) cases, Y chromosome testing can reveal the number of men (from different paternal lines) whose DNA is being detected and whether the defendant’s Y chromosome is consistent with one of these paternal lines.291 Because only males have Y chromosomes, the female DNA in a mixture has no effect.
Finally, rather than extracting DNA from a mixture of cells and performing PCR-CE on the resulting mixture of DNA molecules, it may be possible to isolate individual cells and analyze the DNA one cell at a time,292 either by pushing
289. Peter Gill, as quoted in John M. Butler, Forensic DNA Typing: Biology, Technology, and Genetics of STR Markers 166 (2d ed. 2005).
290. The nucleus of a sperm cell lies behind a protective structure that does not break down as easily as the membrane in an epithelial cell. This makes it possible to disrupt the male and female epithelial cells first and remove their DNA, and then to use a harsher treatment to disrupt the sperm cells. Heather E. McKiernan & Phillip B. Danielson, Molecular Diagnostic Applications in Forensic Science, in Molecular Diagnostics 371 (George P. Patrinos ed., 3d ed. 2017). Other methods of differential extraction also have been studied or applied to forensic samples. E.g., Lana Ostojic et al., Micromanipulation of Single Cells and Fingerprints for Forensic Identification, 51 Forensic Sci. Int’l: Genetics 102430 (2021), https://doi.org/10.1016/j.fsigen.2020.102430.
291. E.g., State v. Polizzi, 924 So. 2d 303, 308–09 (La. Ct. App. 2006) (testing for Y-STRs on “the genital swab with the DNA profile from the Defendant’s buccal swab, . . . the Defendant or any of his paternal relatives could not be excluded as having been a donor to the sample from the victim,” while “99.7 percent of the Caucasian population, 99.8 percent of the African American population, and 99.3 percent of the Hispanic population could be excluded as donors of the DNA in the sample”); State v. Bander, 208 P.3d 1242, 1250 (Wash. Ct. App. 2009) (Y-STR profiling showed that “99.9 percent of the population could be excluded as possible contributors to the sample extracted from the electrical cords recovered from Gardner’s body”).
292. See Jianye Ge et al., Precision DNA Mixture Interpretation with Single-Cell Profiling, 12 Genes 1649 (2021), https://doi.org/10.3390/genes12111649.
PCR-CE to its limits293 or by employing the newer sequencing methods sketched in the section above titled “Sequencing methods.”294 Without physical separation, however, the multiple genotypes in the sample can produce mixed profiles. Analysts then have to interpret these more complex patterns in terms of the individual genotypes that might be present. Inferring the probable genotypes that are combined in a mixed profile is known as mixture deconvolution. This interpretive process is described in the next subsection.
What Is Mixture Deconvolution?
The goal of mixture deconvolution is twofold: (1) to deduce the individual genotypes that are intertwined in an electropherogram that contains peaks from the DNA of different individuals; and (2) to assess the significance of the fact that a defendant has some or all of such a genotype.295 This analysis is computational. A simplified example illustrates the nature of the procedure that has been in use for over 20 years.296 Suppose we create a mixture by adding equal, easily detectable amounts of DNA from two unrelated individuals and type it at just one autosomal STR locus. Let us say that the contributors are each heterozygotes—each individual has two alleles per locus—and that all the alleles are distinct. The electropherogram will have four peaks of roughly equal heights at locations that we can designate A, B, C, and D. An analyst who is given this electropherogram without any information about the contributors could reason as follows:
293. E.g., Kaitlin Huffman et al., Y-STR Mixture Deconvolution by Single Cell Analysis, 68 J. Forensic Sci. 275 (2023), https://doi.org/10.1111/1556-4029.15150 (individual Y-STR profiles recovered from direct PCR amplification of single-cell subsamples from two- and three-person mixtures).
294. See also section titled “Did the Sample Contain Enough DNA?” and note 112 above; cf. Lucie Kulhankova et al., Single-Cell Transcriptome Sequencing Allows Genetic Separation, Characterization and Identification of Individuals in Multi-Person Biological Mixtures, 6 Communications Biology 201 (2023), https://doi.org/10.1038/s42003-023-04557-z (initial study using single-cell RNA transcript sequences and other genetic data to successfully deconvolute laboratory-created mixtures of blood from 2- to 9-person mixtures in ratios as low as 1 to 60).
295. Some courts have held mixture interpretations admissible without any statistical assessment. E.g., Rodriguez v. State, 273 P.3d 845, 851–52 (Nev. 2012); People v. Her, 157 Cal. Rptr. 3d 40 (Cal. Ct. App. 2013).
296. Tim M. Clayton et al., Analysis and Interpretation of Mixed Forensic Stains Using DNA STR Profiling, 91 Forensic Sci. Int’l 55 (1998), https://doi.org/10.1016/s0379-0738(97)00175; Ian W. Evett et al., Taking Account of Peak Areas When Interpreting Mixed DNA Profiles, 43 J. Forensic Sci. 62 (1998); Peter Gill et al., Interpreting Simple STR Mixtures Using Allele Peak Areas, 91 Forensic Sci. Int’l 41 (1998), https://doi.org/10.1016/s0379-0738(97)00174-6.
The minimum number of contributors is two, but there could be more. The four peaks are of equal height, so if there are only two contributors to the mixture, their genotypes could be any of the following six combinations:
CONTRIBUTOR 1 CONTRIBUTOR 2 AB CD AC BD AD BC CD AB BD AC BC AD
These can be written more compactly using a slash to separate the two contributors’ genotypes and listing them as AB/CD, AC/BD, etc. There cannot be exactly three contributors (unless one of them did not contribute enough DNA to have any impact on the peak heights).297 If there are four contributors, the genotypes could be AB/CD/AB/CD, AC/BD/AC/BD, AD/BC/AD/BC, AA/BB/CC/DD, AC/BC/AB/DD, AC/BC/AD/BD, AC/AD/CD/BB, or BC/BD/CD/AA and more (rearranging the order to assign the two-allele genotypes to the different contributors 1 through 4). As five or more contributors are considered, it gets still more complicated.
If the mixed, two-contributor sample had markedly different proportions of DNA, there could be a pattern of tall and short peaks across multiple loci (as in Figure 6), suggesting that the short ones represent the DNA of a “minor contributor” and the tall ones come from the DNA of a “major contributor.” By using the peak heights and locations, the analyst might be able to “deconvolute” the pattern of peaks into the most likely underlying genotypes and then see if the defendant has one of these genotypes.298 Great effort has gone into formulating a systematic procedure for determining the number of contributors to consider,299 to listing all possible sets of genotypes that could give rise to a mixed STR profile, to scratching some off the list as inconsistent with the peak-height information, and to performing a statistical assessment of an inclusion of a defendant who has one of the genotypes left on the list. The standard algorithm for this “manual” deconvolution has six or seven steps.300 The process requires the
297. For instance, if the three genotypes were AB/CD/AB, the A and B peaks would be twice the height of the C and D peaks, but they are not.
298. See, e.g., Roberts v. United States, 916 A.2d 922, 932–35 (D.C. 2007) (holding such inferences to be admissible).
299. Decisions or assumptions about the number of contributors could be assisted by other evidence about the events that resulted in the mixture.
300. For summaries of the steps, see Jo-Anne Bright & Michael D. Coble, Forensic DNA Profiling: A Practical Guide to Assigning Likelihood Ratios 7–15 (2020); Butler et al., supra note 105; see also Frederick R. Bieber et al., Evaluation of Forensic DNA Mixture Evidence: Protocol for Evaluation, Interpretation, and Statistical Calculations Using the Combined Probability of Inclusion, 17 BMC Genetics 125
analyst to make mostly binary decisions about which peaks are alleles as opposed to artifacts (stutter peaks, pull-up, dye blobs).301
It is now generally recognized that to avoid an “expectation effect” on the analyst’s judgments, the allele calls should be made before considering the genotypes of any suspects or other possible contributors to the mixed sample. Then the analyst must decide which genotypes are included or excluded from the mixture. The task becomes even more difficult when the quantity of the DNA from some contributor is marginal, with the possibility of some peaks that would be expected for larger amounts of DNA dropping out to leave only a partial profile.302 Mixtures of degraded or inhibited DNA also are harder to interpret because parts of individual profiles may be missing. Nevertheless, it has been said that “[t]he binary method served the forensic community well for a number of years; however it was mostly restricted to single-source, two-person, and some three-person mixtures (e.g., those with a clear major component).”303 Other observers saw manual mixture interpretation as erratic,304 with some laboratories
(2016), https://doi.org/10.1186/s12863-016-0429-7; Peter Gill et al., DNA Commission of the International Society of Forensic Genetics: Recommendations on the Interpretation of Mixtures, 160 Forensic Sci. Int’l 90 (2006), https://doi.org/10.1016/j.forsclint.2006.04.009; Scientific Working Group on DNA Analysis Methods (SWGDAM), SWGDAM Interpretation Guidelines for Autosomal STR Typing by Forensic DNA Testing Laboratories (Jan. 12, 2017, rev. July 13, 2021), https://perma.cc/2ZCW-Q6L6.
301. On these artifacts, see supra section titled “Capillary electrophoresis and fluorescence.”
302. On drop-out, see supra section titled “Did the Sample Contain Enough DNA?” For a detailed analysis of the manual interpretation steps in the context of an example with these complications, see Michael D. Coble, Worked Mixture Example, in Butler, supra note 113, at 537.
303. Bright & Coble, supra note 300, at 15.
304. An oft-cited but limited study is Itiel E. Dror & Greg Hampikian, Subjectivity and Bias in Forensic DNA Mixture Interpretation, 51 Sci. & Just. 204 (2011), https://doi.org/10.1016/j.sci-jus.2011.08.004. It reports aggregated conclusions of (1) two analysts in Georgia who compared a complex four- or five-man mixture in a gang-rape case with the profiles of three suspects, and (2) 17 analysts at an unnamed laboratory given, as an abstract exercise, the mixed profile and the profile of just one of the three suspects. The two groups reached different conclusions. The extent to which the reported differences prove much about the impact of biasing information and the reproducibility of manual mixture deconvolution in general is discussed in David H. Kaye, The Design of “The First Experimental Study Exploring DNA Interpretation,” 52 Sci. & Just. 126 (2012), https://doi.org/10.1016/j.scijus.2011.10.003, and Itiel E. Dror, Cognitive Forensics and Experimental Research About Bias in Forensic Casework, 52 Sci. & Just. 128 (2012), https://doi.org/10.1016/j.scijus.2012.03.006. More systematic interlaboratory comparisons of up to five-person mixtures from 1997 through 2018 are tabulated in John M. Butler et al., DNA Mixture Interpretation: A NIST Scientific Foundation Review 80–81 (NISTIR 8351-DRAFT, June 2021) (tbl. 4.8), https://doi.org/10.6028/NIST.IR.8351-draft. The reasons for variability in the reports from participants in two U.S. interlaboratory studies are outlined in John M. Butler et al., NIST Interlaboratory Studies Involving DNA Mixtures (MIX05 and MIX13): Variation Observed and Lessons Learned, 37 Forensic Sci. Int’l: Genetics 80, 81 (2018), https://doi.org/10.1016/j.fsigen.2018.07.024; cf. Michael Coble et al., Analysis of Forensic Mixtures, in Forensic DNA Analysis in Criminal Investigations and Humanitarian Disasters 49, 58–59 (Henry Ehrlich et al. eds., 2020) (“Studies from European laboratories [citations omitted] have typically shown wide variation in the results within and between
performing the task poorly.305 Despite the degree to which “subjective” judgment can go into the determination of which peaks are real, which are artifacts, which are masked, and which are absent for some other reason, courts generally have rejected arguments that manual mixture analysis is so unreliable or so open to manipulation that the results are inadmissible.306
What Statistical Assessment of the Comparison to a Defendant’s Profile Has Been Conducted?
When manual deconvolution of a mixed profile is complete and a defendant’s profile is compatible with it, most laboratories will attempt to supply a statistical assessment to indicate how powerful the evidence is.307 Methods for doing so fall
the participants’ laboratories. [In] two interdisciplinary studies in 2005 and 2013 . . . involving laboratories performing STR genotyping in the United States and Canada . . . overall results were concordant with the studies from Europe. . . .”). Proficiency tests, which sometimes include two- or three-person mixtures, are listed in Butler et al., DNA Mixture Interpretation: A Scientific Review, supra, at 78–79 (tbl. 4.7) (reporting small error rates); see also Todd Bille et al., Study of CTS DNA Proficiency Tests with Regard to DNA Mixture Interpretation: A NIST Scientific Foundation Review, 13 Genes 2171 (2022), https://doi.org/10.3390/genes13112171 (re-analyzing the proficiency test data and concluding that “[s]ample switching, contamination, and reporting results incorrectly are serious errors [but not a consequence of] the mixture interpretation strategy”).
305. For example, an inquiry into mixture interpretations at the Washington, D.C., forensic science laboratory at the behest of the U.S. Attorney’s Office for the District of Columbia led to the ouster of the laboratory’s management. See supra note 129. The common errors that were identified mostly pertained to the statistical interpretation of inclusions in mixture cases. Barber v. United States, 179 A.3d 883, 892 n.17 (D.C. 2018). Other jurisdictions have issued letters to convicted defendants notifying them that older calculations of the combined probability of inclusion (see infra section titled “(1) Combined Probability of Inclusion (CPI) and Exclusion (CPE)”) might be inaccurate. Skinner v. State, 484 S.W.3d 434 (Tex. Crim. App. 2016); Rosado v. State, 267 So.3d 4 (Fla. Dist. Ct. App. 2019) (“The notice stated that the Broward Sheriff’s Office Crime Laboratory inappropriately used Combined Probability of Inclusion (CPI) to calculate the statistical probabilities of genetic profiles in complex mixture DNA cases.”); Tex. Forensic Sci. Comm’n, Summary of Texas CPI Case Review Process, Sept. 15, 2016, https://perma.cc/8MAS-EURJ.
306. United States v. Carney, No. 1:20-cr-121, 2022 WL 1155902 (S.D. Ohio Apr. 19, 2022) (reasoning that manual deconvolution for three-person mixtures satisfies Daubert largely because it could be tested and was in widespread use and is still in use for some mixtures in 20% of laboratories); State v. Garland, 942 N.W.2d 732 (Minn. 2020); Roberts v. United States, 916 A.2d 922, 932 n.9 (D.C. 2007) (citing cases).
307. But see supra note 295 (on admissibility without a statistical assessment). The SWGDAM guidelines require a quantitative statement of some kind. SWGDAM, supra note 300, § 3.2.1 (“Except for a reasonably assumed contributor, the laboratory shall perform statistical analysis in support of any inclusion (or a ‘cannot be excluded’ conclusion) irrespective of the number of alleles detected and the quantitative value of the statistical analysis.”).
into two classes: inclusion–exclusion probabilities and likelihood ratios. The latter have greater support within the forensic DNA science community.308
Probability of inclusion or exclusion
Combined probability of inclusion (CPI) and exclusion (CPE).
At first glance, it might seem that it is easy to describe the significance of a match to a mixed profile. We could simply ask what fraction of the population has genotypes that are not excluded by the alleles in the mixed profile. The answer in no way depends on the profiles of the victim, suspect, or person of interest; furthermore, no assumptions or inferences about the number of contributors are necessary for the calculation. For example, suppose we are sure that at one locus, the mixed profile indicates three alleles—A, B, and C, and no others—with population proportions 0.1, 0.2, and 0.3, respectively. The single-locus genotypes of people who could not be excluded as contributing to the profile are AA, BB, CC, AB, AC, and BC. Their (Hardy-Weinberg) probabilities are 0.12 + 0.22 + 0.32 + 2(0.1)(0.2) + 2(0.1)(0.3) + 2(0.2)(0.3) = 0.36.309 So 36% of the population could not be excluded at this locus. Performing the same calculation at the other loci and multiplying them (linkage equilibrium) gives the “combined probability of inclusion,”310 often referred to as CPI or RMNE (for “random man not excluded,” although its use is not restricted to male contributors). The combined probability of exclusion (CPE) is 1 minus the CPI.
Despite its computational simplicity,311 CPI has been deprecated as “hard to justify”312 and an “interim” measure.313 In the three-allele mixture, for example,
308. See infra section titled “Likelihood ratios (LRs).”
309. See supra note 169.
310. Adjustments for population structure (see supra section titled “Could an Unrelated Person Be the Source?”) can be applied. Examples of how to compute CPI in different scenarios are given in SWGDAM, supra note 300, § 4C.
311. Determining the set of loci and alleles to use in the computation can be anything but simple. On the subtleties in using CPI properly, see Barber v. United States, 179 A.3d 883, 892 n.17 (D.C. 2018); Bieber et al., supra note 300; Coble et al., supra note 264, at 243–44; SWGDAM, supra note 300, § 4C.
312. NRC II, supra note 7, at 130.
313. Bieber et al., supra note 300, at 3. PCAST, supra note 105, at 82, expressed skepticism of the “foundational validity” of CPI, and stressed that “DNA analysis of complex mixtures should move rapidly to more appropriate methods based on probabilistic genotyping.” The group added that “[i]f, for a limited time, courts choose to admit results based on the application of CPI, validity as applied would require that, at a minimum, they be consistent with the rules specified in the paper.”
it counts pairs of people who could not jointly be the two contributors.314 Thus, it does not actually resolve the mixture profile into the genotypes of those individuals who probably contributed to the mixed sample. Moreover, it disregards peak-height information, and it ignores the fact that there may be a known contributor such as the victim.
The number of reported opinions responding to challenges to CPI as lacking scientific validity or general acceptance is relatively small. Some courts have reasoned that the CPI is no different than the random-match probability accepted in single-source cases.315 More often, courts recognize that the CPI has been criticized in scientific literature but conclude that it remains generally accepted as a reasonable or often conservative way to express the significance of an inclusion.316
Random-match probability (RMP).
Specifying the number of contributors in the mixture allows the expert to compute a somewhat different probability of inclusion, called the random-match probability (RMP). The RMP also can use peak heights and other information to deconvolve the mixed profile in light of any known or assumed contributor profiles. After taking all these things into account, the analyst will be left with a reduced set of plausible genotypes of the contributors. The RMP is just the sum of the probabilities for each of these genotypes. For example, suppose the victim’s account and other information in a rape case establishes that one man committed the rape, and the victim’s genotype at a locus is AB. DNA from a vaginal swab demonstrates the presence of three alleles A, B, and C with the peak for A and B being about the same height and C being much lower. The male contributor’s genotype could be AC, BC, or CC,317
314. For instance, the genotype pairs AA/AA, AA/BB, AA/CC, AA/AB, and AA/AC can be ruled out. They can produce no more than two peaks in the mixture electropherogram.
315. E.g., People v. Sandifer, 65 N.E.3d 969, 980 (Ill. App. Ct. 2016); cf. Coy v. Renico, 414 F. Supp. 2d 744, 762 (E.D. Mich. 2006) (as “mere[] extensions of the generally accepted and generally reliable techniques applied to single source DNA samples,” CPI and CPE satisfy due process).
316. E.g., State v. Bander, 208 P.3d 1242 (Wash. Ct. App. 2009); cf. State v. Garland, 942 N.W.2d 732, 744 (Minn. 2020) (relying on unspecified “evidence before the district court . . . including a peer-reviewed scientific article regarding best practices for using CPE” to conclude that “CPE has long been accepted by the forensic community as a valid method for calculating the probability of exclusion from a DNA mixture”).
317. The rapist must be responsible for the small peak C. Because it is small, the man is a minor contributor to the sample. A genotype of CC would explain the pattern of major peaks for A and B and a minor peak for C. Genotypes AC and BC for the rapist also would explain this pattern. For a small quantity of DNA of type AC, the A peak gets boosted slightly but remains approximately equal to the B peak. For a minor contribution of genotype BC, the B peak gets boosted, but only slightly. Because no other male genotypes (AA, AB, and BB) can explain the minor peak C in the mixture profile, the rapist must be genotype AC, BC, or CC, as stated in the text.
and the expert could estimate the frequency of these genotypes (using the population allele frequencies posited in the previous subsection) as 2(0.1)(0.3) + 2(0.2) (0.3) + (0.3)(0.3) = 0.27. Instead of reporting the 36% for the single-locus inclusion probability as in the CPI calculation, the expert could report a smaller inclusion probability of 27%.318
Testimony about the RMP (or the frequency with which unrelated individuals would match the deduced, smaller set of genotypes) is often encountered.319 Sometimes the RMP or frequency is described as if it were the CPI.320 Issues likely to arise with the RMP are whether identification of major and minor alleles is correct and how the number of contributors was determined.
Likelihood ratio (LR)
Definition of the likelihood ratio.
Rather than testify to inclusion probabilities for close relatives or for randomly selected, unrelated members of the suspect population, a DNA expert can present a likelihood ratio (LR) to express the evidentiary value of a DNA comparison.321 In the most general formulation,
318. More examples and more refined calculations are in SWGDAM, supra note 300, § 4A; see also Bright & Coble, supra note 300, at 12–14. In two-person mixture cases in which it is reasonable to assume that the victim’s DNA is present, the other contributor’s genotype might be clear from the remaining peaks, and the RMP will be the same as in a single-source case for that contributor.
319. E.g., United States v. Carney, No. 1:20-cr-121, 2022 WL 1155920 (S.D. Ohio Apr. 19, 2022) (RMP for the major contributor to a three-person mixture was said to be “1 in 299 septillion 400 sextillion” and therefore dispositive even though no alleles from the minor contributors could be reported); United States v. Graves, 465 F. Supp. 2d 450, 453 (E.D. Pa. 2006) (RMPs of 1/2, 1/2,900, and 1/3,600); People v. Roberts, 283 Cal. Rptr. 3d 357 (Cal. Ct. App. 2021); State v. Gonzalez, 204 A.3d 1183, 1189 (Conn. App. Ct. 2019); State v. Lowery, 427 P.3d 865 (Kan. 2018); State v. Hannon, 703 N.W.2d 498, 508–09 (Minn. 2005).
320. E.g., People v. Brown, 82 N.E.3d 148, 169–70 (Ill. App. Ct. 2017) (The state’s expert “testified that based on the only 6-loci analysis, he could determine only that defendant could not be excluded as a contributor to the major profile. . . . [T]he frequency which estimated the chance a random person would be included as a contributor in that major DNA profile [was] 1 in 670,000 black, 1 in 580,000 white, or 1 in 6.1 million Hispanic unrelated individuals. . . . The DNA Advisory Board has endorsed two methods . . . in cases of mixed DNA samples: (1) the combined probability of inclusion (or its reverse, the combined probability of exclusion) or (2) the likelihood ratio calculation.”). It is not always clear from an opinion whether the expert is giving a CPI or an RMP. E.g., State v. Dwyer, 985 A.2d 469, 478–79 (Me. 2009) (giving “inclusion probabilities” without indicating whether they were CPIs or RMPs). As a result, some of the opinions cited above as illustrative of one method or the other could be misclassified.
321. In civil and criminal cases that involve DNA testing for parentage, LRs are routinely introduced under the name “paternity index.” 1 McCormick on Evidence, supra note 1, § 211.
an LR is the ratio between (1) the probability of the observed data when one hypothesis about the origin of the data is true and (2) the probability of the data when a non-overlapping hypothesis is true.322 In symbols,
where the vertical line stands for “given” or “conditional on.” For DNA evidence, H1 could be a “contributor hypothesis” that names the defendant as the contributor or one of the contributors, and H2 could be a “noncontributor hypothesis” that names individuals other than the defendant as the contributors. When multiple contributors are likely, various pairs of hypotheses may need to be considered to avoid misstating the value of the evidence.323
The baseline for understanding the value of the LR is 1—the ratio when the probabilities are equal. Equal evidence probabilities mean that the evidence is not probative of which hypothesis is true. For example, if H1 asserts that a defendant’s DNA is in a trace-evidence sample, and H2 asserts that the defendant’s identical twin’s DNA is in the sample, then LR = 1. The DNA data have the same probability under each scenario and therefore are not probative of which twin is a contributor to the sample.
The value of the LR normally would be different if the noncontributor hypothesis H2 were that a different relative or an unrelated person is a contributor. Consider a single-source sample from the crime scene: If the STR profile of the sample is more probable if the defendant’s DNA is in it than if the alternative person’s DNA is there, then the LR exceeds 1. The profile data are more compatible with the defendant’s being the source (H1) than with the alternative (H2). As such, the data can be said to support H1 (compared to H2). Conversely, if the LR is less than 1, the data support H2.
In the legal literature, LRs often are presented as a way to quantify probative value.324 The idea is that relevant evidence changes the probability of some material fact, and the probative value of this evidence is the extent of the change. As noted in “Appendix: Conditional Probability and Bayes’ Rule” of the Reference Guide on Statistics and Research Methods, in this manual, under certain conditions, the change in the odds (known to statisticians as the Bayes’ factor) is simply the LR. When this is the case, the odds after receiving the evidence (the posterior odds) are just the odds before receiving the evidence times the LR:
posterior odds = LR × prior odds
322. Mathematically, when the variable is essentially continuous (such as the height of a peak on an electropherogram) rather than discrete (such as the number of tandem sequences in an STR allele deduced from a peak), the LR is a ratio of probability densities. The definition above also omits a proportionality constant in both the numerator and denominator that cancels out when the ratio is formed.
323. Richard Wivell et al., An Investigation into Compound Likelihood Ratios for Forensic DNA Mixtures, 14 Genes 714 (2023), https://doi.org/10.3390/genes14030714.
324. See 1 McCormick on Evidence, supra note 1, § 185.
This version of Bayes’ rule reinforces the idea that evidence with an LR of 1 has no probative value. Multiplying by 1 has no effect on the odds of H1 relative to H2.
LRs are versatile. With DNA evidence, they can be used to convey the probative value of an inclusion for complex, mixed samples as well as for single-source samples with respect to a variety of alternative explanations for the composition of the sample. For instance, the hypothesis advanced by the prosecution might be that the “defendant and his deceased wife . . . were contributors to a DNA mixture profile drawn from a blood stain on defendant’s sweatshirt,” and the contrasting hypothesis could be that DNA from “two randomly selected individuals” was in the stain.325
Moreover, LRs do not require a binary, include-exclude determination. They do not even require analytical and stochastic thresholds for allele determinations. As such, they can reflect more of the information in the electropherograms than both the CPI and the RMP can. Many forensic geneticists and statisticians regard them as particularly advantageous for mixture interpretation,326 and laboratories across the world have turned to probabilistic-genotyping software to produce LRs to interpret electropherograms from low-template, degraded, or mixed DNA samples with greater consistency than unassisted human judgment can provide.327
In evaluating this development, it is important to keep in mind that the LR is merely a mathematical expression. The utility of any stated value for the quantity depends on how it was computed. Are the hypotheses or propositions appropriate to the case at hand? This is a question of relevance. Has the computational system been shown to give numbers that properly discriminate between the propositions for the DNA samples in the case? This is a question of validity. Assuming validity, how should LRs be presented to lay factfinders so that they
325. People v. Ramsaran, 79 N.E.3d 1120 (N.Y. App. Div. 2017).
326. E.g., Bright & Coble, supra note 300, at 11 (“The LR is our preferred approach to interpreting forensic DNA mixtures.”); Royal Soc’y, Forensic DNA Analysis: A Primer for Courts 36 (2017) (“Likelihood ratios are generally accepted as being the most appropriate method for evaluating the evidential strength of DNA profiles.”); Peter Gill et al., DNA Commission of the International Society for Forensic Genetics: Assessing the Value of Forensic Biological Evidence—Guidelines Highlighting the Importance of Propositions: Part I: Evaluation of DNA Profiling Comparisons Given (Sub-) Source Propositions, 36 Forensic Sci. Int’l: Genetics 189 (2018), https://doi.org/10.1016/j.fsigen.2018.07.003; Peter Gill et al., DNA Commission of the International Society of Forensic Genetics: Recommendations on the Evaluation of STR Typing Results that May Include Drop-out and/or Drop-in Using Probabilistic Methods, 6 Forensic Sci. Int’l: Genetics 679, 684 (2012), https://doi.org/10.1016/j.fsigen.2012.06.002 (“probabilistic approaches and likelihood ratio principles are superior to classical methods”). United Kingdom guidelines regard statements of LRs as the only acceptable approach to mixture interpretation. Forensic Science Regulator, Guidance: DNA Mixture Interpretation (FSR-G-222 Issue 3) (2020), https://perma.cc/L34N-Y54X.
327. For a concise history of the transition to probabilistic genotyping systems, see Michael D. Coble & Jo-Anne Bright, Probabilistic Genotyping Software: An Overview, 38 Forensic Sci. Int’l: Genetics, 219, 219–21 (2019), https://doi.org/10.1016/j.fsigen.2018.11.009.
fairly convey the weight of the evidence with respect to the relevant hypotheses? This is a question of probative value and prejudice. The remaining subsections elaborate on these points and describe the ways in which LRs for DNA evidence are computed.
Binary genotyping.
Traditional forensic STR-CE profiling is “binary” or “deterministic.” Like a baseball umpire calling a pitch a strike or a ball, an analyst (or software) labels a peak as either an allele or not. An allele call requires that the peak height exceed an analytical threshold and that the peak not look like an artifact.328 At each locus, the analyst decides whether there is one allele (homozygosity) or two different alleles (heterozygosity) on the pair of paternal and maternal chromosomes. If the reported profiles in two samples are consistent, there is a profile match. At that point, taking the reported matching profile as 100% certain, analysts can quote LRs for inclusions as the reciprocal of the RMP.329 Thus, the data from the laboratory instruments are processed in two phases—a binary match/no-match phase, and a separate phase to give an LR for the categorical match (by regarding the analyst’s inclusion of the defendant as an adequate summary of the data).
This two-stage procedure works well enough in many cases (and in the absence of better alternatives). The 1996 NRC committee report endorsed then-available binary LRs (based solely on the positions of DNA fragments subjected to electrophoresis) as opposed to the CPI.330 SWGDAM promulgated guidelines for calculating these LRs.331 Despite objections based on Frye or Daubert, appellate courts that have considered binary LRs in mixture cases have upheld their admission.332
328. See supra section titled “Miniaturization for rapid capillary electrophoresis” n.52 (before Table 5); supra section titled “Did the Sample Contain Enough DNA?”
329. E.g., Augillard v. Madura, 257 S.W.3d 494, 498–99 (Tex. Ct. App. 2008) (“the test showed a complete match at all seventeen DNA markers with a likelihood ratio exceeding one trillion”). The LR is the reciprocal of the RMP if the match, as determined by thresholds and human judgment, has probability 1 when H1 is true (if the defendant is a source, the analyst is certain to report an inclusion) and probability RMP when H2 is true (if a random, unrelated person is a source, the probability of inclusion is just the random-match probability).
330. NRC II, supra note 7, at 129 (“when the contributors to a mixture are not known or cannot otherwise be distinguished, a likelihood-ratio approach offers a clear advantage and is particularly suitable”).
331. SWGDAM, supra note 300, § 4(B).
332. E.g., State v. Garcia, 3 P.3d 999 (Ariz. Ct. App. 1999) (applying Frye); Commonwealth v. Gaynor, 820 N.E.2d 233, 252 (Mass. 2005); People v. Coy, 669 N.W.2d 831, 835–39 (Mich. Ct. App. 2003) (incorrectly treating mixed-sample likelihood ratios as a part of the statistics on single-source DNA matches that had already been held to be generally accepted); cf. Coy v. Renico, 414 F. Supp. 2d 744, 762–63 (E.D. Mich. 2006) (likelihood ratio for a mixed stain in People v. Coy, supra, was consistent with due process).
Probabilistic genotyping software (PGS).
Binary LRs have significant limitations. Using only the genotypes as reported from the initial match/no-match phase overlooks any uncertainty in the allele calls and related single-locus genotype determinations. In particular, it ignores the possibility of allelic dropout or drop-in, which could make the assignment of possible genotypes less than certain. It tosses out any alleles that might be producing peak heights below the laboratory’s analytical threshold (or that are wrongly thought to be artifacts).333 In effect, a peak above the line is treated as if it has probability 1 for being an allele, while a peak just below the line is treated as if it has probability zero of being an allele. But surely, if an allele is present (or absent) in a sample, then the probability of a peak just below the threshold is not radically different from a peak just above the line. Newer, probabilistic genotyping systems overcome these limitations by computing a likelihood ratio P(data|H1) ÷ P(data|H2) without having to declare a match or inclusion.
They do so by using weighted averages, for each hypothesis, of the probabilities that all sets of genotypes that could be in the complicated mixture profile are in fact there—based on how often the possible genotypes occur in the relevant population.334 These “prior probabilities” are weighted by the probability that the genotype set (if present) would give rise to the pattern in the electropherogram.335 Thus, a possible genotype set that is more likely to give rise to the electropherogram if it is assumed to be in the mixture profile gets more weight.336
Over the past two decades, researchers have developed a variety of PGS programs with increasingly complete statistical models of the production of true peaks and artifacts in electropherograms. The simplest programs (called qualitative, discrete, or semi-continuous) consider only the alleles that might be
333. See, e.g., Butler, supra note 113, at 39–40.
334. For example, under a contributor hypothesis H1, such as “the defendant and a brother are the source of the semen,” there might be just one set of genotypes at a given locus that conceivably could have produced the electropherogram. For concreteness, let’s say this set is AB/AC, meaning that one brother is AB and the other is AC. Under a noncontributor hypothesis H2 of two random, unrelated men, there might be two sets, such as AB/AC and AA/BC. The prior probability of each of these genotype sets comes from population-genetic models and allele-frequency databases.
335. The genotypes-to-data probability, P(data|genotypes), depends entirely on the sample and the CE measurement process. It is the same under the contributor and the noncontributor hypotheses.
336. The mathematical expression for the weighted averaging is in Peter Gill et al., A Review of Probabilistic Genotyping Systems: EuroForMix, DNAStatistX and STRmix™, 12 Genes 1559, at 2 (2021), https://doi.org/10.3390/genes12101559, and Mark W. Perlin & Alexander Sinelnikov, An Information Gap in DNA Evidence Interpretation, 4 PLoS ONE e8327, at 2 (2009), https://perma.cc/RZ3T-KLHV.
present337 and do not model the height of the peaks.338 They incorporate estimated probabilities of allelic drop-in and drop-out339 when assigning weights on the prior probabilities.340 The output is an LR that draws on more of the information in the mixture electropherogram.341 One such program, the Forensic Statistical Tool developed at the New York City Office of the Chief Medical Examiner, was the subject of considerable litigation342 and is no longer used.343
These programs have been supplanted by “continuous” or “quantitative” systems that model peak heights themselves, automatically accounting for drop-in and drop-out as well as stutter peaks, degradation, and other complicating factors. Two types of continuous systems have been developed. One type posits an explicit mathematical function relating to peak heights. It uses a common statistical method for estimating parameters and applies the estimates to find the genotypes-to-data probabilities.344 The other type relies on Bayes’ rule to obtain these weights.345
337. Even in this regard, many “semi-continuous methods do not model artefacts such as stutter. In these models, peaks must be assigned as stutter or labelled as allelic by an analyst prior to interpretation [by software].” Jo-Anne Bright et al., A Series of Recommended Tests When Validating Probabilistic DNA Profile Interpretation Software, 14 Forensic Sci. Int’l: Genetics 125, 126 (2015), https://doi.org/10.1016/j.fsigen.2014.09.019.
338. At most, they use peak heights in more limited ways, “to inform the probability of dropout or to infer a major donor genotype by applying different drop-out probabilities per contributor.” Nevertheless, “[t]hese systems do represent an advance over the binary model as they can take account of multiple contributors, low-template DNA and replicated samples.” Gill et al., supra note 336, at 3.
339. Drop-in and drop-out probabilities come from experiments on how often peaks from samples with known alleles disappear (drop below the analytical threshold) or appear when the known samples do not contain known alleles for those peaks. See Coble & Bright, supra note 327, at 221.
340. For a numerical example of how drop-in and drop-out probabilities are incorporated, see Michael Coble et al., Mixtures and Probabilistic Genotyping, in Forensic DNA Applications: An Interdisciplinary Perspective 155, 164–66 (Dragan Primorac & Moses Schanfield eds., 2d ed. 2023).
341. For details on the operation of a leading qualitative system, see Gill et al., supra note 36, at 129–80 (on LRmix and LRmix Studio); see also Bright & Coble, supra note 300, at 107–40.
342. Compare, e.g., United States v. Jones, 965 F.3d 149, 162 (2d Cir. 2020) (“no abuse of discretion in the district court’s conclusion [of] reliability sufficient to support admission” of an LR likelihood ratio of “1,340, showing very strong support”), with State v. Rochat, 269 A.3d 1177 (N.J. App. Div. 2022) (FST not generally accepted).
343. Jones, 965 F.2d at 152.
344. On the general nature of parameters in a statistical model and their estimation, see David H. Kaye & Hal S. Stern, Reference Guide on Statistics and Research Methods, in this manual. In one widely used program, EuroForMix, the statistical model for differences between estimated and expected peak heights is known as the gamma (γ) distribution. Gill et al., supra note 36, at 199–200; Gill et al., supra note 336, at 13–14. The parameters of this distribution are closely related to the proportion of mixture DNA from each contributor and the coefficient of variation of peak heights across all the loci. Gill et al., supra note 36, at 201–05. The parameter values are estimated by the maximum-likelihood method. Id. at 461.
345. Gill et al., supra note 36, at 462. Because peak height is a continuous variable, the version of Bayes’ rule for discrete variables presented in the Reference Guide on Statistics and Research Methods,
The two programs that are most commonly used in the United States are of the latter type. They have the brand names TrueAllele and STRmix. To approximate the genotypes-to-data probabilities, they take a kind of random walk through the space of all possible parameter values,346 spending more time at those combinations at which the pattern of peaks computed by the model come closest to the observed values.347 The relative time spent at each such combination determines the weights P(data|genotype set).348 The technical name for this probabilistic approximation method is Markov chain Monte Carlo (MCMC).349 The “Monte Carlo” part refers to computer simulation of the outcomes of any process that has some randomness in it.350 Simulations rarely give exactly
in this manual, needs to be restated with a probability density function and integration instead of summation. The underlying concepts are the same, however, and, for ease of presentation, this reference guide has not distinguished between discrete probability distributions and probability density functions. The LR for continuous models is a ratio of probability densities.
346. For partial descriptions of the parameters in the peak-height model of STRmix, see Bright & Coble, supra note 300, at 143 (“For the simplest profile . . . these are 1. DNA amount (template) for each contributor to the profile 2. The degradation rate for each contributor to the profile 3. Amplification efficiencies for each locus within the profile.”); Coble et al., supra note 340, at 167 (“template quantity, degradation, stutter ratios, etc.”).
347. At least, that is the expectation. The game of “hot and cold,” in which a person searching for an object in a room is told whether he is getting closer to or farther from the object as he moves around the room is sometimes offered as a metaphor for the computer-simulation procedure. E.g., United States v. Gissantaner, 417 F. Supp. 3d 857 (W.D. Mich. 2019), rev’d, 990 F.3d 457 (6th Cir. 2021); United States v. Lewis, 442 F. Supp. 3d 1122, 1142 (D. Minn. 2020) (Rep. & Recommendation) (extending the analogy to multiple rooms). A better (but still imperfect) analogy might be trying to climb the highest mountain in a region with no visibility by taking proposed steps that increase the elevation with a higher probability than steps that do not, until no proposed step increases the altitude. This should get the climber to the summit if the mountain is smooth and if there are no smaller mountains in the region. Unlike the hot-and-cold game in which one knows when the object is found, in the latter landscape, there is a risk of being fooled into thinking that a smaller peak is the highest there is.
348. See Coble et al., supra note 340, at 167–68.
349. The “Markov chain” part refers to situations in which the next value of a random variable depends to some degree on just the previous value (like a peculiar coin that is a little more likely to turn up heads when tossed if a head had turned up on the previous toss). “Since their invention [in 1905], Markov chains have become extremely important in a huge number of fields such as biology, game theory, finance, machine learning, and statistical physics.” Joseph K. Blitzstein & Jessica Hwang, Introduction to Probability 459 (2015). Nonetheless, the success of an MCMC algorithm in one domain or type of problem does not guarantee that it will work as well in another situation.
350. For example, if we wanted to estimate the probability of a lucky streak of ten heads when tossing a fair coin, we could program a computer to pick a 0 or a 1 at random ten times and see if the sequence was 1111111111. One such simulation would not tell us much, but if the computer simulated ten tosses one million times, keeping track of how many trial sequences were 1111111111, the proportion usually would be close to the true value of the probability. That the inventors of the computer-simulation method referred to a famous gaming casino in naming it is an historical accident. See Nicholas Metropolis, The Beginning of the Monte Carlo Method, Los Alamos Sci., Special Issue 125, 127 (1987), available at https://perma.cc/M53S-Y9X9.
identical estimates if they are rerun, and “[a] simulation result is easy to criticize: how do you know you ran it long enough? How do you know your result is close to the truth? How close is ‘close?’”351
These are questions to be addressed in validation studies, considered in the next subsection. Proof of validity also needs to attend to the more fundamental issue of the adequacy of the parametric models for predicting the observed data from the possible genotype sets. PGS programs that use Bayesian methods and MCMC computations may not use exactly the same statistical models, and one program may require more preliminary input from the DNA analyst or laboratory using it than another.352 Rather than a blind acceptance of the output, the reasonableness and impact of these choices may need to be examined.
Validation of PGS programs.
In the courtroom, the most important output of PGS is the LR for a set of hypotheses that include the defendant as a contributor to the mixture (or to a single-source sample, typically of very low quantity).353 The LR is not a measured physical or chemical property, like height or weight. Rather, it is an expression of the degree to which the electropherogram is more indicative of scenarios that include the defendant as a contributor than it is of scenarios that do not. How, then, do scientists who have written the software convince other scientists that the values of the LR are believable?
Validation has at least three components. First, the mathematical equations used in the software and the ways of solving those equations must be appropriate.354
351. Blitzstein & Hwang, supra note 349, at 421 (also noting that “[t]he simulations may need to run for a vast, unknown amount of time to get decent answers”).
352. See Daniels v. State, 312 So.3d 926 (Fla. Dist. Ct. App. 2021) (whereas STRmix requires laboratory-specific values from “internal validation” studies, TrueAllele does not). TrueAllele also dispenses with analytic thresholds; but STRmix uses them. William C. Thompson, Uncertainty in Probabilistic Genotyping of Low Template DNA: A Case Study Comparing STRMix™ and TrueAllele™, J. Forensic Sci. (2023), https://doi.org/10.1111/1556-4029.15225. However, STRmix may be moving away from these rigid cutoffs. See Duncan Taylor & John Buckleton, Combining Artificial Neural Network Classification with Fully Continuous Probabilistic Genotyping to Remove the Need for an Analytical Threshold and Electropherogram Reading, 62 Forensic Sci. Int’l: Genetics 102787 (2023), https://doi.org/10.1016/j.fsigen.2022.102787.
353. Some PGS programs are also useful for estimating the number of contributors and mixture ratios and for producing a list of high-LR contributor genotypes that could be submitted to an offender DNA database.
354. The conceptual underpinnings of the two main continuous PGS systems have not attracted major criticism. The weighted averaging described in the section titled “Probabilistic genotyping software (PGS)” comes out of the basic mathematics of probability theory and Bayes’ rule. The values for the prior probabilities are population genotype frequencies that are calculated from population-genetics models and are discussed in the section titled “Inference, Statistics, and Population Genetics in Human Nuclear DNA Testing” above. The modeling of the genotypes-to-data probabilities also
In litigation, the general mathematical structure behind most PGS software has not been seriously questioned.355
Second, the coding of the mathematical formulas and techniques for solving the equations must be correct. Examining parts of the source code of a program and testing whether modules work as they are supposed to as the program is written and revised can reveal coding and logical problems.356 Thus, voluntary quality-assurance standards for software (and hardware) “verification and validation” are well known among software engineers.357 These standards are sometimes cited to support motions for discovery of all of the source code of a PGS program or to buttress an argument that the program cannot be found to be scientifically valid or generally accepted without publishing the code (open-access software), or at least affording defense experts extensive review of proprietary software, perhaps under a confidentiality order.358
needs to be sound. Finally, the statistical procedures (like an MCMC algorithm, which is not itself the genetic or statistical model at the heart of PGS) must be capable of solving (at least approximately) the equations to which they are applied.
355. E.g., United States v. Lewis, 442 F. Supp. 3d 1122, 1145 (D. Minn. 2020) (Rep. & Recommendation) (“The underlying principles on which STRmix is based—the MCMC, the Hastings-Metropolis algorithm, and Bayesian statistics—are not themselves challenged as unreliable. Nor is the purely mathematic calculation of relative probabilities (i.e., the likelihood ratio).”); People v. Davis, 290 Cal. Rptr. 3d 661, 679 (Cal. Ct. App. 2022) (“[t]he scientific and mathematical principles behind STRmix are well-established and widely-accepted in the scientific community”); People v. Wakefield, 195 N.E.3d 19, 28 (N.Y. 2022) (“It was undisputed that the foundational mathematical principles [of TrueAllele] (MCMC and Bayes’ theorem) are widely accepted in the scientific community.”). But see United States v. Gissantaner, 417 F. Supp. 3d 857, 870 (W.D. Mich. 2019), rev’d, 990 F.3d 457 (6th Cir. 2021) (suggesting that software that implements Bayes’ rule is problematic because “Bayesian reasoning ‘breaks down in situations where information must be conveyed from one person to another such as in courtroom testimony’”).
356. “Black box” testing, which involves no inspection of the underlying code, is also a standard procedure. See, e.g., Christopher Henard et al., Comparing White-Box and Black-Box Test Prioritization, ICSE ’16: Proceedings of the 38th International Conference on Software Engineering 523 (2016), https://doi.org/10.1145/2884781.2884791.
357. In this context, “verification” refers to making sure that the software does what the specifications call for. “Validation” refers to ensuring that the product meets the needs of its users. The ISO/IEC/IEEE 29119 series of standards offer guidance on “V & V” for business and other environments. See ISO/IEC/IEEE 29119–1:2022(en) (Software and systems engineering—Software testing—Part 1: General concepts). They go beyond establishing scientific or technical merit.
358. See, e.g., People v. Wakefield, 195 N.E.3d 19 (N.Y. 2022); State v. Watkins, 648 S.W.3d 235 (Tenn. Ct. Crim. App. 2021); cf. Nathaniel Adams et al., Letter to the Editor—Appropriate Standards for Verification and Validation of Probabilistic Genotyping Systems, 63 J. Forensic Sci. 339 (2018), https://doi.org/10.1111/1556-4029.13687 (urging developers to utilize the IEEE V & V standards). Without necessarily relying on the V & V software engineering standards, legal academics tend to advocate disclosure of source code. See Rebecca Wexler, Life, Liberty, and Trade Secrets: Intellectual Property in the Criminal Justice System, 70 Stan. L. Rev. 1343, 1376 (2018) (citing articles). The authors of some PGS programs are more skeptical of the need for defendants’ experts or third parties to peruse the code itself. E.g., Duncan A. Taylor et al., Commentary: A “Source” of
The third component of validation is direct testing of the PGS by presenting “a wide range of evidence types, representing a typical case, as well as extreme examples.”359 The performance on data from a large number of mixtures of known composition can provide convincing empirical proof of the validity of a conceptually sound system (for mixtures like those in the experiment). Mixed samples can be constructed in the laboratory by combining DNA in varying ratios and total amounts from known individuals.360 Mixed profiles for validation also can be simulated by combining known single-sample-profile data together mathematically.361 Yet a third approach uses samples from cases in which defendants were convicted.362 The idea in each approach is to assemble two sets of paired data for testing the PGS: contributor-mixture pairs and
Error: Computer Code, Criminal Defendants, and the Constitution, 8 Frontiers Genetics 33 (2017), https://doi.org/10.3389/fgene.2017.00033 (maintaining that it is “nearly impossible to identify subtle errors in code by viewing the code”).
A different argument about PGS source code is that studying it is somehow like cross-examining an “artificial intelligence,” and the Confrontation Clause therefore gives the defendant in a criminal case the right to see the code itself when the AI’s output is testimonial under Crawford v. Washington, 541 U.S. 36 (2004). Wakefield, 195 N.E.3d at 44–45 (concurring opinion). A more straightforward constitutional analysis would look to the Due Process Clause and ask whether the need to inspect the source code is critical in light of other methods of testing the operation of the software.
359. Gill et al., supra note 336, at 10.
360. Samples also can be made from DNA with different degrees of degradation or other complicating factors.
361. Artificially generating the validation profiles rather than profiling “made-up samples” has been advocated to “control the input variables exactly and produce profiles where the expected answer is known. [I]t is effectively impossible to create an exact 1:1 [or other mixture-ratio] mixture in vitro, [whereas artificially mixed profiles] are simply tidy single source, major, minor and balanced profiles.” Bright et al., supra note 337, at 126.
362. Treating these defendants as true contributors gives us a set of presumed-contributor-mixture pairs for testing the PGS. A much larger set of known noncontributor-mixture pairs for testing can be created by substituting genotypes of the defendants in all the other (unrelated) cases for the actual defendant. For a study employing this design, see Mark W. Perlin et al., New York State TrueAllele® Casework Validation Study, 58 J. Forensic Sci. 1458, 1469 (2013), https://doi.org/10.1111/1556-4029.12223; cf. David H. Kaye et al., Validating the Probability of Paternity, 31 Transfusion 823 (1991), https://doi.org/10.1046/j.1537-2995.1991.31992094670.x (examining the empirical distribution of the likelihood ratio for HLA-typing results in civil paternity cases). One might object that the presumed-contributor-mixture pairs could include some falsely convicted defendants. Obviously, it would be better to restrict the true-pair group to only true (contributor-mixture) pairs, as can be done with manufactured mixtures. However, a fraction of noncontributor-mixture pairs in the presumed-contributor-mixture pairs can only make it harder to find that a valid PGS typically generates large LRs for the group designated as true pairs. A clear difference in PGS performance, as between the two large groups of pairs, is thus still good evidence of validity. A failure to find a difference is more ambiguous. It could be explained as either a lack of validity or as a consequence of having too many falsely convicted offenders’ profiles in the true-pairs group.
noncontributor-mixture pairs.363 Each method of forming the two sets has advantages and disadvantages, and a series of studies with the different designs is best.
The PGS can be applied to these validation test sets in several ways. Analysts can verify that the program gives large LRs for true pairs in simple cases (ones they can readily resolve without the software).364 As the information available from the mixture declines (because of more complex peaks and low-template samples), the LR values should drop as well. Beyond such rudimentary checks, rates of misleading LRs can be determined.365 The observed proportion of cases in the validation test set for which the PGS LR points in the wrong direction (toward exclusion for contributors or toward inclusion for noncontributors)366 is a kind of “error rate” for the PGS.367
Nonetheless, these misleading-LR rates do not tell the whole story. Even if the overall rates of misleading evidence are small, how do we know that an LR of 1 million is dramatically more powerful than an LR of, say, 1,000 or 100?368 To address this question, validation studies can examine whether larger LRs are indeed associated with smaller rates of misleading evidence. Do LRs of 1,000 or
363. The false-pair cases can be formed by pairing the data for each mixture with many randomly generated genotypes (using the allele frequencies in any population of interest).
364. Bright et al., supra note 337, at 126.
365. See Richard Royall, On the Probability of Observing Misleading Statistical Evidence, 95 J. Am. Stat. Ass’n 760 (2000).
366. The tendency of the computed LRs to exceed 1 for mixtures that include the defendant’s DNA has been called “sensitivity.” The tendency to be less than 1 for mixtures that do include the defendant has been called “specificity.” E.g., Scientific Working Group on DNA Analysis Methods (SWGDAM), Guidelines for the Validation of Probabilistic Genotyping Systems § 21 (2015), accessible via https://perma.cc/5V7F-6VJ7. The terms are an extension of their more established meaning for binary tests. See supra section titled “Validity.”
367. United States v. Gissantaner, 990 F.3d 457, 465 (6th Cir. 2021); Colin Aitken & Franco Taroni, The History of Forensic Inference and Statistics: A Thematic Perspective, in Handbook of Forensic Statistics 3, 23 (David Banks et al. eds., 2021); David H. Kaye, Forensic Statistics in the Courtroom, in id. at 225. Other measures of performance also are in use. See Aitken & Taroni, supra, at 23–24; Daniel Ramos et al., Validation of Forensic Automatic Likelihood Methods, in Handbook of Forensic Statistics 143 (David Banks et al. eds., 2021). Repeatability of computed LRs (also referred to as “precision” and “reproducibility” in the standards and writing on PGS) can be studied by rerunning the PGS on the same data. It is an issue for MCMC computations. See, e.g., David W. Bauer et al., Validating TrueAllele Interpretation of DNA Mixtures Containing up to Ten Unknown Contributors, 65 J. Forensic Sci. 380 (2020), https://doi.org/10.1111/1556-4029.14204.
368. The court in United States v. Lewis, 442 F. Supp. 3d 1122 (D. Minn. 2020), suggested that proof that “[t]he variation in the precise LR numbers generated by STRmix from one run to the next is very small” (deviating by no more than a factor of ten) answered this question. Id. at 1130. But replicability must not be mistaken for validity. Generating similar results on repeated tries is not necessarily the same as generating correct results. A scale that always reported objects with true weights of 1 gram and 1 kilogram as weighing 1–10 grams (first object) and 100–1,000 kilograms (second object) would massively overstate the weight of the heavier object.
more crop up in the noncontributor-mixture cases more than one time in 1,000? Do LRs of 1 million or more arise more often than one time in a million in these cases? If not, the PGS LRs are well calibrated (or at least not systematically overstated).369
The point at which enough well-designed studies have been conducted to answer these questions is not easily defined. Simply counting the number of studies with the word “validity” in their title only scratches the surface. Did the studies include large numbers of contributor and noncontributor pairs? Which performance metrics were used? Is the case within the range of conditions covered in the studies? In 2016, the President’s Council of Advisors on Science and Technology reported that PGS programs “clearly represent a major improvement over purely subjective interpretation,”370 but that the studies to that point, which came from the software developers rather than from completely separate researchers,371 “have adequately explored only a limited range of mixture types (with respect to number of contributors, ratio of minor contributors, and total amount of DNA).”372 Additional studies of as many as 10-person mixtures followed.373 Almost invariably, courts have upheld the admissibility of
369. Duncan Taylor et al., Testing Likelihood Ratios Produced from Complex DNA Profiles, 16 Forensic Sci. Int’l: Genetics 165 (2015), https://doi.org/10.1016/j.fsigen.2015.01.008. Simulating enough noncontributor pairs to test LRs of very large magnitudes can be challenging. For one strategy, see Duncan Taylor et al., Importance Sampling Allows HdTrue Tests of Highly Discriminating DNA Profiles, 27 Forensic Sci. Int’l: Genetics 74 (2017), https://doi.org/10.1016/j.fsigen.2016.12.004.
370. PCAST, supra note 105, at 79.
371. The report evinced concern over the involvement of the developers of the software in the validity studies of their products: “Appropriate evaluation of the proposed methods should consist of studies by multiple groups, not associated with the software developers, that investigate the performance and define the limitations of programs by testing them on a wide range of mixtures with different properties.” Id. (emphasis in original). The issue of developer involvement is discussed in Gissantaner, 990 F.3d 457 (6th Cir. 2021); United States v. Lewis, 442 F. Supp. 3d 1122, 1147–49 (D. Minn. 2020) (Rep. & Recommendation); People v. Wakefield, 195 N.E.3d 19 (N.Y. 2022); People v. Williams, 147 N.E.3d 1131 (N.Y. 2020); and other cases.
372. PCAST, supra note 105, at 80. The report added that “[t]he two most widely used methods (STRMix and TrueAllele) appear to be reliable within a certain range, based on the available evidence and the inherent difficulty of the problem. Specifically, these methods appear to be reliable for three-person mixtures in which the minor contributor constitutes at least 20 percent of the intact DNA in the mixture and in which the DNA amount exceeds the minimum level required for the method.” Id.
373. E.g., Michael S. Adamowicz et al., Internal Validation of MaSTR™ Probabilistic Genotyping Software for the Interpretation of 2–5 Person Mixed DNA Profiles, 13 Genes 1429 (2022), https://doi.org/10.3390/genes13081429; Bauer et al., supra note 367 (TrueAllele); Jo-Anne Bright et al., Internal Validation of STRmix™—A Multi-Laboratory Response to PCAST, 34 Forensic Sci. Int’l: Genetics 11 (2018), https://doi.org/10.1016/j.fsigen.2018.01.003; Sara Riman et al., Examining Performance and Likelihood Ratios for Two Likelihood Ratio Systems Using the PROVEDIt Dataset, 16 PLoS ONE e0256714 (2021), https://doi.org/10.1371/journal.pone.0256714 (EuroForMix and STRmix applied
PGS results in the face of objections to their scientific validity or general scientific acceptance,374 although some cases have been remanded for more complete hearings.375
Presentation of LRs.
Whether likelihood ratios result from categorical matching or from PGS programs, courts may need to consider the manner in which the ratios are presented at trial. The legal concerns are avoiding overstating what science has to offer (an aspect of Rules 403 and 702) and preventing unfair prejudice or confusion (Rule 403). Like random-match probabilities, LRs are easily transposed,376 and it can be argued that large numbers might be overvalued.377 With random-match probabilities, we saw that courts have reasoned that the possibility of transposition does not justify a blanket rule of exclusion.378 Appellate courts have not addressed the issue for LRs,379 but it is not obvious
to the same mixture profiles). But in 2021, a small group of scientists and other personnel working at NIST released a draft of a “scientific foundation review” that stated “[c]urrently, there is not enough publicly available data to enable an external and independent assessment of the degree of reliability of DNA mixture interpretation practices, including the use of probabilistic genotyping software (PGS) systems.” Butler et al., supra note 105, at 6, 75.
374. E.g., Gissantaner, 990 F.3d 457 (STRmix); Lewis, 442 F. Supp. 3d 1122 (STRmix); People v. Davis, 290 Cal. Rptr. 3d 661 (Cal. Ct. App. 2022) (STRmix); State v. Simmer, 935 N.W.2d 167 (Neb. 2019) (TrueAllele); Wakefield, 195 N.E.3d 19 (TrueAllele); cf. United States v. Williams, 382 F. Supp. 3d 928 (N.D. Cal. 2019) (LR inadmissible because “Bullet” PGS program was only validated for up to four-person mixtures, and it could not be shown that the mixture in question did not come from five individuals).
375. E.g., People v. Wortham, 180 N.E.3d 516 (N.Y. 2021) (New York City Office of the Chief Medical Examiner’s program, FST).
376. For a few examples, see People v. Pike, 53 N.E.3d 147, 167 (Ill. App. Ct. 2016) (“estimates how much more likely it is that the suspect is the source of the evidence than it is that the evidence originated from a randomly selected member of the population unrelated to the suspect”); State v. Pickett, 246 A.3d 279 (N.J. App. 2021) (“a statistic measuring the probability that a given individual was a contributor to the sample against the probability that another, unrelated individual was the contributor”); Commonwealth v. Treiber, 121 A.3d 435, 445 (Pa. 2015) (“the hair was 1,000 times more likely to have come from appellant’s dog than any other dog”). These statements mischaracterize the likelihood ratio as the relative probability of a source hypothesis given the DNA data rather than the relative probability of the data given the hypotheses. Kaye et al., supra note 193, § 14.2.2.
377. Some commentary maintains that likelihood ratios are inherently prejudicial. E.g., William C. Thompson, DNA Evidence in the O.J. Simpson Trial, 67 U. Colo. L. Rev. 827, 855–56 (1996); see also Richard C. Lewontin, Population Genetic Issues in the Forensic Use of DNA, in 1 Modern Scientific Evidence: The Law and Science of Expert Testimony § 17–5.0, at 703–05 (David L. Faigman et al. eds., 1st ed. 1998).
378. See section titled “Frequencies, Probabilities, and Prejudice” above.
379. Trial court rulings admitting likelihood ratios over the unfair prejudice objection were upheld in State v. Ayers, 68 P.3d 768, 778 (Mont. 2003), and State v. Watkins, 648 S.W.3 235, 265 (Tenn. Crim. App. 2021).
why the rule would be different.380 The usual expectation is that “[a] district court concerned that the jury might misunderstand what the likelihood ratio means could require advocates to describe it in a way that will not generate unfair prejudice or mislead the jury.”381
But what would that be? It has been suggested that experts should cease characterizing LRs as statements that “a match is X times more/less probable than a coincidence,”382 for that formulation seems to invite the misunderstanding that the LR is the odds in favor of the contributor hypothesis. Better explanations of the LR would clarify that the value pertains to the probability of the evidence (the STR data) rather than the probability of the conclusion (who the contributors are). An LR of x means that the DNA data are x times more likely if the hypothesis that includes the defendant as a contributor is correct than if the hypothesis that only other people are contributors is correct. One textbook for practitioners advises that “[a]n example of suitable phrasing (where the LR = one billion) is ‘The evidence is a billion times more likely if the person of interest is a contributor than if a random, unrelated person is a contributor.’”383
Even with the correct conditional phrasing, however, the number may be puzzling. Factfinders are not used to thinking about ratios of probabilities. To help, experts could offer analogies. One such analogy is a partial fingerprint pattern that one in a thousand people would possess. A DNA comparison with
380. Psychological research to ascertain the effects of likelihood-ratio statements, as opposed to other quantitative and qualitative indicators of probative value, does not demonstrate that research subjects overvalue the evidence as described in questionnaires. See Busey, supra note 186; Edward K. Cheng, The Burden of Proof and the Presentation of Forensic Results, 130 Harv. L. Rev. F. 154, 161 (2017) (“An increasingly complex literature has emerged on lay understanding of likelihood ratios and how such quantitative information is best presented. Research thus far has yielded no easy answers . . . .”); Heidi Eldridge, Juror Comprehension of Forensic Expert Testimony: A Literature Review and Gap Analysis, 1 Forensic Sci. Int’l: Synergy 24 (2019), https://doi.org/10.1016/j.fsisyn.2019.03.001; Martire, supra note 186. For some studies in this area, see Brandon L. Garrett et al., Error Rates, Likelihood Ratios, and Jury Evaluation of Forensic Evidence, 65 J. Forensic Sci. 1199 (2020), https://doi.org/10.1111/1556-4029.14323; William C. Thompson et al., Perceived Strength of Forensic Scientists’ Reporting Statements About Source Conclusions, 17 L. Probability & Risk 133 (2018), https://doi.org/10.1093/lpr.mgy012; William C. Thompson & Eryn J. Newman, Lay Understanding of Forensic Statistics: Evaluation of Random Match Probabilities, Likelihood Ratios, and Verbal Equivalents, 39 L. & Hum. Behav. 332 (2015), https://doi.org/10.1037/lhb0000134.
381. United States v. Gissantaner, 990 F.3d 457, 470 (6th Cir. 2021) (internal quotation marks and alteration omitted).
382. Thompson, supra note 352; cf. Eur. Network of Forensic Sci. Inst., Best Practice Manual for Human Forensic Biology and DNA Profiling 29 (ENFSI-DNA-BPM-03, Ver. 01. 2022) (“Caution is required when using the word ‘match’ in statements because it might imply ‘identity’. The expert avoids any verbal statement that might imply that he/she is making an opinion on the identity of the questioned DNA.”).
383. Bright & Coble, supra note 300, at 30.
an LR of 1,000 has the same probative value as the partial print even though the phenomena have nothing to do with each other.384 Experts also could describe an LR as a measure of support for a scenario and add a qualitative expression such as “weak,” “strong,” “very strong,” and so on for the degree of support.385 Forensic statisticians originally proposed “verbal scales” for less objectively determined LRs for all kinds of forensic-science identification evidence, from fingerprints to toolmarks to handwriting, in the hope that these scales would encourage greater standardization in expressing the strength of the evidence and better comprehension of LRs.386 The Department of Justice’s Uniform Language for Testimony and Reports for PGS presents one set of verbal tags: uninformative (for LR = 1), limited support (2 to <100), moderate support (100 to <10,000), strong support (10,000 to <1,000,000), and very strong support (≥1,000,000).387 Analysts are allowed but not required to use these tags (and no others) along with the actual value.388 If any tag is used, then the examiner must list the full scale “to provide context for the numerical value.”389
384. Other analogies are possible. For example, an LR of about 1,000 is what you would get if someone flipped one of two coins—either a normal, evenly balanced coin or a trick coin with heads on both sides. You do not know which coin was flipped, but the evidence is that it came up heads every time in ten tosses. The evidence—the ten heads—is about 1,000 times more probable if it was the trick coin that was tossed than if it was the normal one. The expert would have to add that the probability that the trick coin was tossed also would depend on how likely it is that the person doing the flipping would choose the trick coin instead of the normal one—a factor that is beyond the data from the experiment on the coin (tossing it ten times). Similarly, the expert would have to say that she cannot give the probability that the defendant’s DNA was present. She can only report the probability if it was present compared to the probability if the alternative she considered was what had happened.
385. E.g., United States v. Williams, 382 F. Supp. 3d 928, 934–35 (N.D. Cal. 2019) (“Based on SERI’s [Serological Research Institute’s] verbal scale, this result indicated ‘very strong’ support for the proposition that Elmore is a contributor to the sample.”) (footnote omitted).
386. See, e.g., Raymond Marquis et al., Discussion on How to Implement a Verbal Scale in a Forensic Laboratory: Benefits, Pitfalls and Suggestions to Avoid Misunderstandings, 56 Sci. & Just. 364 (2016), https://doi.org/10.1016/j.scijus.2016.05.009.
387. U.S. Dep’t of Just., Uniform Language for Testimony and Reports for Forensic Autosomal DNA Examinations Using Probabilistic Genotyping Systems (Sept. 13, 2022) (link at https://perma.cc/8VFG-KMHB).
388. Id. at 3.
389. Id. The ULTR does not explain how dividing the continuum of possible LR values from zero to infinity into arbitrary segments provides meaningful context. As indicated in the section titled “Are Random-Match Probabilities That Are Smaller Than False-Positive Error Probabilities Irrelevant or Prejudicial?” of the third edition of this reference guide (Reference Manual on Scientific Evidence (3d ed. 2011)), some statisticians would argue that a more helpful explanation is to present an LR as the extent to which the evidence changes the odds in favor of a factual claim. See also Kaye & Stern, supra note 12, Appendix C.
DNA and RNA Typing of Bodily Fluids or Tissues
Standard DNA genotyping methods help answer the question of whose DNA is in the sample. In many cases, however, the more critical question is “how” rather than “who.” For example, a defendant accused of rape might concede that there was an assault but argue that the victim’s DNA found near the zipper of his pants resulted from her touching the fabric with her hands and was not from vaginal fluid, as the prosecution proposed.390 Forensic scientists call such factual claims activity-level propositions.391 To supply activity-level evidence, molecular biology tests for differentiating between different types of bodily fluids and tissues are beginning to be used in casework.392 Although the sequence of base pairs in DNA throughout the body is essentially identical, different parts of the coding DNA are active in each type of specialized cell that forms a tissue. Gene “expression” occurs when part of the double-stranded DNA molecule is transcribed into messenger RNA and that mRNA is translated into a protein.393 Some “housekeeping” proteins are in all tissues, but other proteins—and the associated mRNA transcripts—vary markedly in the extent to which they are present in different tissues. Skin cells have a different expression pattern than
390. Cf. Holtzclaw v. State, 448 P.3d 1134, 1148 (Okla. Ct. Crim. App. 2019) (no error for prosecutor to argue that DNA near the defendant’s zipper was “transferred by the vaginal fluids” even though the DNA analyst did not make that inference); Chong Wang et al., Body Fluid Identification by Messenger RNA Profiling in Sexual Assault, 8 J. Forensic Sci. & Med. 118 (2022), https://doi.org/10.4103/jfsm.jfsm_54_21 (“the suspect argued that the condom was just touched by the victim”).
391. R. Cook et al., A Hierarchy of Propositions: Deciding Which Level to Address in Casework, 38 Sci. & Just. 231 (1998), https://doi.org/10.1016/S1355-0306(98)72117-3; Bas Kokshoorn et al., Activity Level DNA Evidence Evaluation: On Propositions Addressing the Actor or the Activity, 278 Forensic Sci. Int’l 115 (2017), https://doi.org/10.1016/j.forsciint.2017.06.029. To address the activity-level questions, forensic-science researchers and practitioners would like to bring to bear, in a standardized and systematic way, scientific studies of transfer, persistence, prevalence, and recovery of DNA. Duncan Taylor & Bas Kokshoorn, Forensic DNA Trace Evidence Interpretation: Activity Level Propositions and Likelihood Ratios (2023); Peter Gill et al., DNA Comm’n of the Int’l Soc’y for Forensic Genetics: Assessing the Value of Forensic Biological Evidence—Guidelines Highlighting the Importance of Propositions. Part II: Evaluation of Biological Traces Considering Activity Level Propositions, 44 Forensic Sci. Int’l: Genetics 102186 (2020), https://doi.org/10.1016/j.fsigen.2019.102186.
392. Andrea Patrizia Salzmann et al., mRNA Profiling of Mock Casework Samples: Results of a FoRNAP Collaborative Exercise, 50 Forensic Sci. Int’l: Genetics 102409 (2021), https://doi.org/10.1016/j.fsigen.2020.102409; Titia Sijen & SallyAnn Harbison, On the Identification of Body Fluids and Tissues: A Crucial Link in the Investigation and Solution of Crime, 12 Genes 1728, § 4.1 (2021), https://doi.org/10.3390/genes12111728 (“RNA typing has been applied regularly at the Netherlands Forensic Institute since 2012”). This section discusses mRNA testing, but the related proteins themselves also can be analyzed with mass spectrometry. See, e.g., Forensic Proteomics: Protein Identification and Profiling (Eric D. Merkley ed., 2019).
393. See section titled “Genes and Gene Products” above.
liver cells, for example.394 Researchers have identified certain mRNA transcripts that are characteristic of forensically relevant fluids and thus can serve as markers for those fluids.
The mRNA in a biological sample can be extracted along with the DNA.395 Using the technologies for PCR amplification and capillary electrophoresis,396 or sequencing397 (see section above titled “What Are DNA Polymorphisms and How Are They Detected?”), the laboratory can try to determine the fluid or tissue in the trace-evidence sample. Other RNA transcripts than mRNA also have been proposed for fluid and tissue identification, as has testing for a chemical change to the DNA molecule that silences gene expression.398
Twins
Identical twins originate from a single fertilized egg cell (a zygote). Instead of multiplying to develop into a single embryo, fetus, and child, the developing set of cells breaks up into two or more masses that grow into separate children.399 Fraternal twins come from two different eggs fertilized by different sperm; those twins have many differences in their genomes. But in monozygotic twins, all the cells of the progeny come from the original zygote with a single genome. To the extent that the original chromosomes are faithfully copied in every cell
394. See Human Protein Atlas, The Tissue Section—Tissue-Based Map of the Human Proteome, https://perma.cc/5UW9-ULHN.
395. Amy D. Roeder & Cordula Haas, Body Fluid Identification Using mRNA Profiling, in Forensic DNA Typing Protocols 13 (William Goodwin ed., 2016), https://doi.org/10.1007/978-1-4939-3597-0.
396. Patricia Pearl Albani & Rachel Fleming, Developmental Validation of an Enhanced mRNA-based Multiplex System for Body Fluid and Cell Type Identification, 59 Sci. & Just. 217 (2019), https://doi.org/10.1016/j.scijus.2019.01.001; Tiffany R. Layne et al., Rapid Microchip Electrophoretic Separation of Novel Transcriptomic Body Fluid Markers for Forensic Fluid Profiling, 13 Micromachines 1657 (2022), https://doi.org/10.3390/mi13101657. To use a PCR-CE system, the mRNA first must be “reverse transcribed” into a DNA segment.
397. Cordula Haas et al., Forensic Transcriptome Analysis Using Massively Parallel Sequencing, 52 Forensic Sci. Int’l: Genetics 102486 (2021), https://doi.org/10.1016/j.fsigen.2021.102486; E. Hanson et al., Messenger RNA Biomarker Signatures for Forensic Body Fluid Identification Revealed by Targeted RNA Sequencing, 34 Forensic Sci. Int’l: Genetics 206 (2018), https://doi.org/10.1016/j.fsigen.2018.02.020.
398. Changes to DNA that do not alter the sequence of nucleotides are called “epigenetic.” The attachment of a small chemical group (a methyl group) on a gene stops it from producing proteins. The pattern of methylation of the DNA is different in different tissues. Athina Vidaki & Manfred Kayser, Recent Progress, Methods and Perspectives in Forensic Epigenetics, 37 Forensic Sci. Int’l: Genetics 180 (2018), https://doi.org/10.1016/j.fsigen.2018.08.008.
399. The monozygotic twinning rate is about 1 pregnancy per 250. Kurt Benirschke, Multiple Gestation: The Biology of Twinning, in Creasy and Resnik’s Maternal-Fetal Medicine: Principles and Practice 53, 53 (Robert K. Creasy et al. eds., 7th ed. 2014).
division, monozygotic twins are genetically identical.400 Forensic STR analysis, which considers only tiny slices of the entire genome, cannot tell one from the other.
This limitation has thwarted prosecutions in which both twins were believed to have committed the crime401 or in which an “evil twin” defense to an alleged crime committed by one twin was offered.402 But variations in observable characteristics detectable by molecular tests related to DNA do exist.403 This section discusses one of them that has made an initial appearance in criminal and civil litigation.404 It has been proffered as “a realistic option, fit for practical forensic casework”405 that can support claims that one identical twin rather than the other is the source of trace-evidence DNA.406
Point mutations accumulate throughout life as cells split, giving rise to more and more distinguishable lines of cells. Even before birth, many “identical”
400. See supra note 25 & accompanying text.
401. In perhaps the most dramatic of these cases, German authorities did not bring charges in a “massive jewelry heist” because they could not decide which twin to charge. The German twins sent a message that they were “proud of the German constitutional state and gave it their thanks.” Twins Suspected in Spectacular Jewelry Heist Set Free, Speigel Online Int’l, Mar. 19, 2009, https://perma.cc/W6D8-GYH9, last visited Mar. 9, 2023.
402. Alison Gee, Twin DNA Test: Why Identical Criminals May No Longer Be Safe, BBC News Mag., Jan. 15, 2014, https://perma.cc/QM7N-DWWM; Richard Willing, Identical Twins Complicate Use of DNA Testing, USA Today, Nov. 30, 2004, at A03.
403. There are differences in which genes are expressing proteins (“epigenetic” differences). Mario F. Fraga et al., Epigenetic Differences Arise During the Lifetime of Monozygotic Twins, 102 Proc. Nat’l Acad. Sci. 10604 (2005), https://doi.org/10.1073/pnas.0500398102. Forensic-science researchers have begun to study how to use this variation on samples of the same type of tissue from two twins. E.g., Benjamin Planterose Jiménez et al., Equivalent DNA Methylation Variation Between Monozygotic Co-Twins and Unrelated Individuals Reveals Universal Epigenetic Inter-Individual Dissimilarity, 22 Genome Biology 18 (2021), https://doi.org/10.1186/s13059-020-02223-9 (“may one day allow universal epigenetic fingerprinting, which for instance is relevant in forensics for differentiating MZ twin individuals”); Athena Vidaki et al., Epigenetic Discrimination of Identical Twins from Blood Under the Forensic Scenario, 31 Forensic Sci. Int’l: Genetics 67 (2017), https://doi.org/10.1016/j.fsigen.2017.07.014. In addition, there are differences in the number of copies of a particular gene present in the genomes of different twins (“copy number variants”). Carl E.G. Bruder et al., Phenotypically Concordant and Discordant Monozygotic Twins Display Different DNA Copy-Number-Variation Profiles, 82 Am. J. Hum. Genetics 763 (2008), https://doi.org/10.1016/j.ajhg.2007.12.011. Furthermore, differences in mitochondrial sequences have been reported. Lijuan Yuan et al., Identification of the Perpetrator Among Identical Twins Using Next Generation Sequencing Technology: A Case Report, 44 Forensic Sci. Int’l: Genetics 102167 (2020), https://doi.org/10.1016/j.fsigen.2019.102167.
404. Burkhart Rolf & Michael Krawczak, The Germlines of Male Monozygotic (MZ) Twins: Very Similar, But Not Identical, 50 Forensic Sci. Int’l: Genetics 102408 (2021), https://doi.org/10.1016/j.fsigen.2020.102408.
405. Michael Krawczak et al., Distinguishing Genetically Between the Germlines of Male Monozygotic Twins, 14 PLoS Genetics e1007756 (2018), https://doi.org/10.1371/journal.pgen.1007756.
406. The technique has been used several times in Europe. Id.; Netherlands Forensic Institute, NFI Reports on Distinguishing Twins Using DNA Analysis, Oct. 19, 2022, https://perma.cc/XWX8-83Y8.
twins differ at some single base pairs (out of billions).407 After the embryo splits into twins, cells in each twin migrate and differentiate into distinct tissue types. If the mutation event occurs after twinning, it will be present only in one twin.408 If it occurs early, before the cells have specialized into all the cell types (nerve cells, blood cells, skin cells, germline cells, and so on), it may be perpetuated in multiple cell types and tissues. And if it occurs farther down the road, it will appear only in the tissue type of the first mutated cell.
To find distinguishing point mutations, a laboratory can perform whole-genome sequencing with an MPS method409 on a sample of blood from each twin. The twins are likely to have a small number of discordant base pairs.410 Knowing the few discrepancies between the twins, the laboratory goes back to the trace-evidence DNA. Suppose the trace-evidence DNA came from a bloodstain. Sanger sequencing of that DNA at the distinguishing SNPs will show which twin’s SNPs are also present in the trace-evidence DNA.
In this example, blood DNA has been compared to blood DNA. Suppose instead that the trace-evidence DNA is from semen, not blood, and the comparison DNA is from swabs of insides of the cheeks (the buccal mucosa) of the twins. The two types of collected cells developed from different parts of the embryo, and this fact complicates the analysis. Two or three weeks after fertilization, a set of cells in the embryo are slated to become germ cells (sperm or ova) in an event known as primordial germ cell specification (PGCS). Whether a mutation is propagated in sperm, in the mucosa, or in both—and whether these outcomes are present in one twin, the other twin, or both—depends on when (relative to PGCS and twinning) and where (within the embryo) they occurred.411 With cross-tissue comparisons, the technique thus requires establishing that at least some of the different SNPs in the twins’ tissues are “twin-specific on the
407. Hakon Jonsson et al., Differences Between Germline Genomes of Monozygotic Twins, 53 Nature Genetics 27, 27 (2021), https://doi.org/10.1038/s41588-020-00755-1 (“monozygotic twins differ on average by 5.2 early developmental mutations and . . . approximately 15% of monozygotic twins have a substantial number of these early developmental mutations specific to one of them”); Rui Li et al., Somatic Point Mutations Occurring Early in Development: A Monozygotic Twin Study, 51 J. Med. Genetics 28 (2014), https://doi.org/10.1136/jmedgenet-2013-101712 (based on different SNPs found in 66 monozygotic twin pairs, “it is likely that each individual carries approximately over 300 postzygotic mutations in the nuclear genome of white blood cells that occurred in the early development”).
408. Conceivably, the same mutation could occur independently after twinning at the very same point within the billions of base pairs in a cell in the other twin. In that case, the single nucleotide change would not distinguish between the twins, but such a duplicated mutation is extremely unlikely.
409. See supra section titled “Sequencing and SNPs.”
410. To prune away sequencing errors with MPS, the laboratory can apply a slower but more accurate method such as Sanger sequencing to the small number of candidate variants.
411. See Jonsson et al., supra note 407.
one hand, but not (too) tissue-specific on the other.”412 In the semen-versus-cheek-swab situation, the premise is that the mutations occurred in early cells that gave rise to both progenitor germ cells and epithelial cells.
The evidentiary issues associated with this method of breaking the STR tie between twins include the validity of the sequencing methods;413 the adequacy of the statistical model for calculating a pertinent likelihood ratio;414 and the laboratory’s correct execution of the sequencing and statistical procedures. Also, a criminal-procedure issue would arise if one or both of the twins refused to give DNA samples. Compelling suspects to give bodily samples (breath, blood, saliva, hair, and fingerprint scrapings) is not unusual,415 and warrants, nontestimonial orders, or subpoenas for DNA samples from third parties have been issued.416 But these cases contemplated forensic STR or VNTR profiling, which reveals limited medical or other socially significant information.417 Whole genome sequencing (WGS) reveals copious genetic information. Even if the only part of the genome that the government cares about and uses is a handful of SNPs that are useful only for differentiating between the two twins, it could be argued that compelling the twins to submit to the extraction of their blood or saliva for WGS is an unreasonable search or seizure.418
412. Rolf & Krawczak, supra note 404. This wrinkle resulted in the exclusion of whole genome sequencing evidence in Commonwealth v. McNair, No. 8414CR10768 (Mass. Suffolk Cnty. Super. Ct., Apr. 11, 2017), pet. for interlocutory rev. denied, No. SJ-2017-0186 (Supreme Jud. Ct., Suffolk Cnty., July 27, 2017). The memorandum opinion is reprinted in David H. Kaye, The Gordian Knot in Commonwealth v. McNair: What Did the Court Decide About Daubert?, Forensic Sci., Stat. & L., Mar. 11, 2019, https://perma.cc/3LJJ-P4K4.
413. See supra section titled “Sequencing methods.”
414. Michael Krawczak et al., Distinguishing Genetically Between the Germlines of Male Monozygotic Twins, 14 PLoS Genetics e1007756 (2018), https://doi.org/10.1371/journal.pgen.1007756; David H. Kaye, Identical Twins, Different Tissues, Disparate DNA, Forensic Sci., Stat. & L., Mar. 7, 2019, https://perma.cc/49AP-Q4WL.
415. Doe v. Senechal, 725 N.E.2d 225 (Mass. 2000) (court-ordered buccal swab to test whether a member of the staff of a residential treatment facility for mentally ill adolescents fathered the child of a patient is a reasonable search and seizure); In re Non-testimonial Identification Order Directed to R.H., 762 A.2d 1239 (Vt. 2000) (saliva sampling by court order based on reasonable suspicion).
416. Bill v. Brewer, 799 F.3d 1295 (9th Cir. 2015) (warrant for police officers’ DNA to exclude them as sources of crime-scene DNA); In re G.B., 139 A.3d 885 (D.C. 2016) (warrant for a buccal swab from the victim of a stabbing for testing of DNA from blood droplets and stains at the apartment where the victim allegedly was stabbed). But cf. Commonwealth v. Kostka, 31 N.E.3d 1116 (Mass. 2015) (order to twin to provide a saliva sample to determine whether he was a fraternal as opposed to an identical twin was unreasonable where there was no showing of how definitive the profiling of a mixed sample from the victim’s fingernail was and no showing that defendant would raise an evil twin defense; and where fingerprint evidence could have excluded even an identical twin).
417. See supra note 39.
418. Cf. section titled “Investigative Genetic Genealogy” below. To forestall arguments over the validity or weight of cross-tissue comparisons, the prosecution in a rape case would need to
Investigative DNA Analysis
Much of this guide has presented DNA analysis as courtroom evidence that a criminal defendant is associated with a crime. The remaining sections describe DNA analysis from the perspective of police who have yet to zero in on a suspect. How can forensic scientists use DNA from a crime scene or a victim as a clue that could lead police to some actual suspects? Computerized databases of DNA profiles serve this function by allowing investigators to compare an unknown profile to millions of profiles from known individuals for “cold hits.” These law-enforcement intelligence databases are the subject of the next section. Police also can take advantage of large databases of other information on individual genomes maintained by companies to help members of the public locate possible relatives. The section below titled “Investigative Genetic Genealogy” describes the use of this genomic data to locate suspects indirectly, by first identifying possible relatives of the unknown source of the crime-scene DNA. A final section considers trace-evidence DNA, not purely as a token of individual identity or a step in constructing a family tree, but rather as an indication of biogeographic origin or external traits.
Offender and Suspect Database Searches
Law-enforcement databases and databanks
In 1989, Virginia became the first state to collect DNA from individuals convicted of certain crimes for a statewide DNA database—a set of records of DNA profiles that could be searched to see whether any of them matched a profile from biological evidence associated with a crime. Other states established similar systems,419 and in 1998, the FBI launched software and hardware—the Combined DNA Index System (CODIS)—to allow nationwide searches to generate
secure semen samples from the twins. The balance of interests that would determine the constitutional reasonableness of compelling a twin to give such a sample would tilt further in the reluctant twin’s favor.
419. Although state and federal statutes authorizing and governing DNA databases for law enforcement are now pervasive, it is not obvious that the laws preclude a local police agency from maintaining its own records of DNA profiles from suspects they encounter or from biological evidence that they collect. “Rapid DNA” instruments and companies marketing software for database management have encouraged this practice. See supra section titled “Miniaturization for rapid capillary electrophoresis.” The “rogue databases” operating outside the constraints and quality assurance requirements of statutorily established systems have sparked concern. Murphy, supra note 287, at 180–88; N.Y. City Bar Ass’n Crim. Courts Comm., Crim. Just. Operations Comm., and Mass Incarceration Task Force, Curbing Unregulated Local DNA Indexing (Apr. 16, 2021).
an extremely valuable investigative lead: the name of an individual whose DNA, very likely, was left at the crime scene.420
CODIS has three layers. At the bottom, local governmental laboratories store the DNA profiles they obtain in a Local DNA Index System (LDIS). In the middle, a State DNA Index System (SDIS) holds these profiles and additional ones generated by statewide laboratories. State law specifies which profiles can be included. A National DNA Index System (NDIS) sits at the top. It houses SDIS profiles that meet federal requirements plus DNA profiles from federal laboratories. Approved law-enforcement laboratories can conduct database searches at any level—local, state, or national.421
These CODIS databases have four main sets of records: a convicted offender index; an arrestee index; a detainee index;422 and a forensic index (of profiles from DNA from crime scenes or left on victims).423 Matches can occur between either the convicted offender, arrestee, or detainee indexes and the forensic index. Cold hits also can occur within the forensic index, linking crimes to each other. The underlying samples (or just the DNA extracts) also are retained, making the system a databank as well as an electronic database.424 Finally, the system as a whole includes “population databases.” These are anonymized research databases used to estimate allele frequencies that go into the equations
420. A pilot project had been started in 1990. U.S. Dep’t of Just., Office of the Inspector General, Combined DNA Index System Operational and Laboratory Vulnerabilities, Audit Report 06–32 (2006). The DNA Identification Act of 1994, 34 U.S.C. §§ 40702–40703 (later amended in various respects), authorized the FBI to create a national database. At the outset of the fully operational system, only nine states participated, and the federal government did not collect DNA from federal offenders. Nathan James, Cong. Reference Serv., DNA Testing in Criminal Justice: Background, Current Law, Grants, and Issues 3 (2014) (CRS Rep. R41800).
421. On the requirements for uploading a crime-scene profile to NDIS so that it will be searched against the offender and arrestee databases, see Cowels v. FBI, 936 F.3d 62 (1st Cir. 2019) (FBI’s refusal to accept a sample that a state court ordered uploaded to NDIS at the request of defendants, who had been convicted but were granted a new trial, was not arbitrary and capricious under the Administrative Procedure Act where it was not clear that the sample was connected to the crime). The scope of the information maintained at each level of the hierarchy of databases varies. LDIS has names and crime information that are not uploaded to SDIS and NDIS.
422. Detainees are “non-United States persons who are detained under the authority of the United States.” 28 C.F.R. § 28.12(b) (2023). “Non-United States persons” means “persons who are not United States citizens and who are not lawfully admitted for permanent residence.” Id. DNA is not routinely collected from “(1) [a]liens lawfully in, or being processed for lawful admission to, the United States; (2) [a]liens held at a port of entry during consideration of admissibility and not subject to further detention or proceedings; or (3) [a]liens held in connection with maritime interdiction.” Id. On the rationale for these exceptions, see 85 Fed. Reg. 13483 (Mar. 9, 2020).
423. It also has profiles from unidentified remains and from samples voluntarily provided by relatives of missing persons. Butler, supra note 43, at 236.
424. Id. at 214–15; United States v. Kriesel, 720 F.3d 1137 (9th Cir. 2013) (describing the reasons for sample retention and holding that Fed. R. Crim. 41(g) does not require the government to return the blood sample after an offender has completed his sentence and term of supervised release).
for genotype frequencies, random-match probabilities, or likelihood ratios.425 They come from other sources and are not searched for cold hits.426
Law-enforcement DNA databases and databanks have provoked considerable litigation. Fourth Amendment challenges to compulsory DNA collection from convicted offenders almost always failed,427 although opinions were divided as to the appropriate doctrinal route around normal warrant and probable cause requirements,428 and several vigorous dissenting opinions emerged.429 Expanding the scope of the databases to include profiles from individuals who were merely indicted or arrested produced a split of authority in the lower courts. Eventually, the Supreme Court, in Maryland v. King,430 narrowly upheld a state statute requiring all custodial arrestees to submit to DNA sampling as part of the booking process for certain violent crimes and burglary.431
425. These quantities are discussed supra in the sections titled “Inference, Statistics, and Population Genetics in Human Nuclear DNA Testing” and “(1) Definition of the likelihood ratio”.
426. Butler, supra note 43, at 215. The failure to recognize the difference between searchable records for possible cold hits and de-identified databases for population frequency research led the state of Texas to destroy its public health system’s repository of newborn blood samples. David H. Kaye, Up in Smoke: 5 Million Neonatal Blood Samples Incinerated, Forensic Sci., Stat. & L., July 27, 2014, https://perma.cc/2LN2-NQXL. On law-enforcement access to newborn samples collected for genetic-disease screening, see Natalie Ram, America’s Hidden National DNA Database, 100 Tex. L. Rev. 1253 (2022).
427. See United States v. Kincade, 379 F.3d 813 (9th Cir. 2004) (en banc); Robin Cheryl Miller, Validity, Construction, and Operation of State DNA Database Statutes, 76 A.L.R. 5th 239 (2000).
428. See Banks v. United States, 490 F.3d 1178, 1183 (10th Cir. 2007) (“our sister circuits have taken different analytical routes to analyzing DNA-indexing statutes” and “our own precedents are divided”); Padgett v. Donald, 401 F.3d 1273, 1278 (11th Cir. 2005) (“Each circuit to address the question has upheld the constitutionality of DNA profiling statutes, but the circuits have disagreed on whether to do so through the special needs analysis or through the traditional balancing test.”).
429. Kincade, 379 F.3d at 842 & 871 (dissenting opinions).
430. 569 U.S. 435 (2013).
431. King has been extended to uphold other database laws that encompass more crimes, that do not automatically remove profiles and samples if no conviction follows the arrest, and that can be used for “familial searching.” Haskell v. Harris, 745 F.3d 1269 (9th Cir. 2014) (en banc) (per curiam); Haskell v. Brown, 317 F. Supp. 3d 1095 (N.D. Cal. 2018); People v. Buza, 413 P.3d 1132 (Cal. 2018) (but defendant’s alleged crime fell within the “serious” category of King); cf. State v. Medina, 102 A.3d 661 (Vt. 2014) (striking down post-arraignment, pre-conviction, DNA sampling under the Vermont constitution). For academic debate on the reach of King and the Fourth Amendment logic of the majority and the dissent, compare Erin Murphy, License, Registration, Cheek Swab: DNA Testing and the Divided Court, 127 Harv. L. Rev. 161 (2013), with David H. Kaye, Maryland v. King: Per Se Unreasonableness, the Golden Rule, and the Future of DNA Databases, 127 Harv. L. Rev. Forum 39 (2013). See also David H. Kaye, Why So Contrived? DNA Databases After Maryland v. King, 104 J. Crim. L. & Criminology 535 (2014); Tracey Maclin, Maryland v. King: Terry v. Ohio Redux, 2013 Sup. Ct. Rev. 359; Andrea Roth, Maryland v. King and the Wonderful, Horrible DNA Revolution in Law Enforcement, 11 Ohio St. J. Crim. L. 295 (2013).
Probative value of database hits
If the DNA profile from a trace-evidence sample matches a profile in an arrestee, detainee, or offender database, the person identified by the cold hit will become the target of the investigation. Prosecution may follow. These database-trawl cases can be contrasted with traditional “confirmation cases” in which the defendant already was a suspect and the DNA testing provided additional evidence. In confirmation cases, statistics such as the estimated frequency of the matching DNA profile in various populations, the equivalent random-match probabilities, or likelihood ratios can be used to indicate the probative value of the DNA match.432
In trawl cases, however, an additional question arises: Does the fact that the defendant was selected for prosecution by trawling require some adjustment to the usual statistics? The legal issues are twofold. First, is a particular quantity—be it the unadjusted random-match probability or some adjusted probability—scientifically valid (or generally accepted) in the case of a database search? If not, it must be excluded under the Daubert (or Frye) standards. Second, is the statistic irrelevant or unduly misleading? If so, it must be excluded under the rules that require all evidence to be relevant and not unfairly prejudicial. To clarify, we summarize the statistical literature on this point. Then, we describe the case law.
The statistical analyses of adjustment.
All statisticians agree that, in principle, the search strategy affects the probative value of a DNA match. One group describes and emphasizes the impact of the database match on the hypothesis that the database does not contain the source of the crime-scene DNA. This is a “frequentist” view. It asks how frequently searches of innocent databases—those for which the true source is someone outside the database—will generate cold hits. From this perspective, trawling is a form of “data mining” that produces a “selection effect” or “ascertainment bias.”433 If we pick a lottery ticket at random, the probability p that we have the winning ticket is negligible. But if we search through all the tickets, sooner or later we will find the winning one. And even if we search through some smaller number N of tickets, the probability of picking a winning ticket is no longer p, but Np.434
432. On the computation and admissibility of such statistics, see supra sections titled “Inference, Statistics, and Population Genetics in Human Nuclear DNA Testing” and “Likelihood ratio (LR)” (in the section titled “Mixtures”).
433. See Kaye & Stern, supra note 12.
434. The analysis of the DNA database search is more complicated than the lottery example suggests. In the simple lottery, there was exactly one winner. The trawl case is closer to a lottery in which we hold a ticket with a winning number, but it might be counterfeit, and we are not sure how many counterfeit copies of the winning ticket were in circulation when we bought our N tickets.
Likewise, if DNA from N innocent people is examined to determine if any of them match the crime-scene DNA, then the probability of a match in this group is not p, but some quantity that could be as large as Np. This type of reasoning led the 1996 NRC committee to recommend that “[w]hen the suspect is found by a search of DNA databases, the random-match probability should be multiplied by N, the number of persons in the database.”435 The 1992 committee436 and the FBI’s former DNA Advisory Board437 took a similar position.
No one questions the mathematics that show that when the database size N is very small compared with the size of the population, Np is an upper bound on the expected frequency with which searches of databases will incriminate innocent individuals when the true source of the crime-scene DNA is not represented in the databases. The Bayesian school of thought, however, suggests that the frequency with which innocent databases will be falsely accused of harboring the source of the crime-scene DNA is basically irrelevant. The question of interest to the legal system is whether the one individual whose database DNA matches the trace-evidence DNA is the source of that trace. As the size of a database approaches that of the entire population, finding one and only one matching individual should be more, not less, convincing evidence against that person. Thus, instead of looking at how surprising it would be to find a match in a large group of innocent suspects, this school of thought asks how much the result of the database search enhances the probability that the individual so identified is the source. The database search is actually more probative than the confirmation search because the DNA evidence in the trawl case is much more extensive. Trawling through large databases excludes millions of people, thereby reducing the number of people who might have left the trace evidence if the suspect did not. This additional information increases the likelihood that the defendant is the source, although the effect is indirect and generally small.438
435. NRC II, supra note 7, at 161 (Recommendation 5.1).
436. The first NRC committee wrote that “[t]he distinction between finding a match between an evidence sample and a suspect sample and finding a match between an evidence sample and one of many entries in a DNA profile databank is important.” It used the same Np formula in a numerical example to show that “[t]he chance of finding a match in the second case is considerably higher, because one . . . fishes through the databank, trying out many hypotheses.” NRC I, supra note 6, at 124. Rather than proposing a statistical adjustment to the match probability, however, that committee recommended using only a few loci in the databank search, then confirming the match with additional loci, and presenting only “the statistical frequency associated with the additional loci.” Id. at 124 tbl. 1.1.
437. DNA Advisory Bd., Statistical and Population Genetics Issues Affecting the Evaluation of the Frequency of Occurrence of DNA Profiles Calculated from Pertinent Population Database(s) (July 2000), https://perma.cc/JQ4C-6ZGR.
438. When the size of the database approaches the size of the entire population, the effect is large. Also, finding that only one individual in a large database has a particular profile raises the probability that this profile is very rare, further enhancing the probative value of the DNA evidence.
Of course, when the cold hit is the only evidence against the defendant, the total package of evidence in the trawl case is less than in the confirmation case. Nonetheless, the Bayesian treatment shows that the DNA part of the total evidence is more powerful in a cold-hit case because this part of the evidence is more complete than when the search for matching DNA is limited to a single suspect. This reasoning suggests that the random-match probability (or, equivalently, the frequency p in the population) understates the probative value of the unique DNA match in the trawl case. And if this is so, then Np is misleadingly large, and the unadjusted random-match probability or frequency p can be used as a conservative indication of the probative value of the finding that, of the many people in the database, only the defendant matches.439
Judicial opinions on adjustment.
The need for an adjustment has been vigorously debated in the statistical literature, and to a lesser extent in the legal literature.440 The dominant view in the journal articles is that the random-match probability, frequency, or likelihood ratio need not be inflated to protect the defendant—at least, not as a matter of mathematics.441 The major opinions to
439. This analysis was developed by David Balding and Peter Donnelly. For informal expositions, see, for example, Peter Donnelly & Richard D. Friedman, DNA Database Searches and the Legal Consumption of Scientific Evidence, 97 Mich. L. Rev. 931 (1999); David H. Kaye, Rounding Up the Usual Suspects: A Legal and Logical Analysis of DNA Database Trawls, 87 N. Car. L. Rev. 425 (2009); Simon Walsh et al., DNA Intelligence Databases, in Forensic DNA Evidence Interpretation 429, 443–44 (John Buckleton et al. eds., 2d ed. 2016); Simon Walsh & John Buckleton, DNA Intelligence Databases, in Forensic DNA Evidence Interpretation 439, 465–68 (John Buckleton et al. eds., 2005). For a related analysis directed at the average probability that an individual identified through a database trawl is the source of a crime-scene DNA sample, see Yun S. Song et al., Average Probability That a “Cold Hit” in a DNA Database Search Results in an Erroneous Attribution, 54 J. Forensic Sci. 22, 23–24 (2009), https://doi.org/10.1111/j.1556-4029.2008.00917.x. Yet another variation is in Ian Ayres & Barry Nalebuff, The Rule of Probabilities: A Practical Approach for Applying Bayes’ Rule to the Analysis of DNA Evidence, 67 Stan. L. Rev. 1447 (2015); cf. John T. Wixted et al., Calculating the Posterior Odds from a Single-match DNA Database Search, 18 L. Probability & Risk 1 (2018), https://doi.org/10.1093/lpr/mgz001 (with commentary).
440. For citations to this literature, see Kaye, supra note 439; Ronald W.J. Meester & Klaas Slooten, DNA Database Matches: A p Versus np Problem, 46 Forensic Sci. Int’l: Genetics 102229 (2020); https://doi.org/10.1016/j.fsigen.2019.102229; Walsh et al., supra note 439, at 443.
441. Marjan J. Sjerps, Probabilistic Considerations when Interpreting Database Searches and Selection Effects, in Handbook of Forensic Statistics 325, 331 (David Banks et al. eds., 2021) (“[N]umerous papers have been published after Donnelly and Friedman [supra note 439] and [A. Philip Dawid, Comment on Stockmarr’s “Likelihood Ratios for Evaluating DNA Evidence When the Suspect Is Found Through a Database Search,” 57 Biometrics 976 (2001)], exploring the controversy with, e.g., Bayesian networks, or rewriting formulas in a more convenient way, reaching for the most part the same conclusion. . . . Thus, from a mathematical point of view, the database search controversy has been solved.”). On the manner and situations in which p, Np, or some combination or variation of the two should be presented, there is less agreement. See ENFSI DNA Working Group, DNA Database
confront this issue agree that p is admissible against the defendant in a trawl case.442 They reason that all the statistics are admissible under Frye and Daubert because there is no controversy over how they are computed. They then assume that both p and Np are logically relevant and not prejudicial in a trawl case.
Kinship trawling (“familial searching”)
Normally, police trawl a DNA database to see if any recorded STR profiles match a crime-scene profile. On occasion, these trawls could lead to charges against a defendant whose DNA is not even in the databank. This can occur for identical twins, one of whom is a convicted offender whose DNA type is on file and the other who has never been typed. If the convicted twin was in prison when the crime under investigation was committed, and if the police realize that he had an identical twin, suspicion should fall on the twin. Presumably, the police would seek a sample from this twin, and at trial it would not be necessary for the prosecution to explain the roundabout process through which the defendant was identified.
In this hypothetical example, the defendant fortuitously was found because a relative’s DNA led the police to him. More generally, the fact that close relatives share more alleles than other members of the same subpopulation can be exploited as a deliberate investigative tool. Rather than search for a match at all the loci in an STR profile, police could search for a near miss—a partial match that is much more probable when the partly matching profile in the database comes from a close relative than when it comes from an unrelated person.443
Management Review and Recommendations (Apr. 2019), accessible via European Network of Forensic Science Institutes (ENFSI) Best Practice Manuals and Forensic Guidelines, https://perma.cc/XN6B-8DAY; David H. Kaye, People v. Nelson: A Tale of Two Statistics, 7 Law, Probability & Risk 249 (2008), https://doi.org/10.1093/lpr/mgn005; Kaye, supra note 439; Meester & Slooten, supra note 440; Murphy, supra note 287, at 118; Sjerps, supra; Wixted et al. (and commentary), supra note 439.
442. People v. Nelson, 185 P.3d 49 (Cal. 2008); United States v. Jenkins, 887 A.2d 1013 (D.C. 2005). The cases are analyzed in Kaye, supra note 439 & note 441. More recent cases are similar. E.g., People v. Turner, 476 P.3d 676 (Cal. 2020); Commonwealth v. Bizanowicz, 945 N.E.2d 356, 362–63 (Mass. 2011). In Turner, the California Supreme Court took a step toward the pure random-match probability approach. 476 P.3d at 693 & 693 n.12 (intimating that the Np statistic would not be “germane” when p is extremely small, but it could be admitted “in some circumstances” when “a profile’s random match probability [is] relatively high”). Why or how to draw this line is not clear.
443. Frederick R. Bieber et al., Finding Criminals Through DNA of Their Relatives, 312 Science 1315 (2006), https://doi.org/10.1126/science.1122655. The FBI distinguishes between kinship trawling to locate an individual outside the database and “partial match” searching to locate an individual inside the database when the crime-scene profile is incomplete. FBI, supra note 9 (questions 25 & 27–32). The latter can incidentally produce a lead to someone outside the database, but it is not an efficient procedure for that purpose.
Analysis of Y-STRs or mtDNA from the DNA sample in the databank then could determine whether the “database inhabitant” probably is in the same paternal or maternal lineage as the unknown individual who left DNA at the scene of the crime. Apparent relatives who emerge from this process become candidates for further investigation based on nongenetic information such as age and location. (Alternatively, a Y-STR or mtDNA database could be queried at the outset for possible matches to the trace-evidence DNA. Several countries have used this strategy.444)
Such “familial searching”445 raises technical and policy questions. The technical and practical challenge is to devise a search strategy that detects the relatives who happen to be in the database while keeping the number of false leads to a tolerable level. For autosomal STR profiling supplemented by Y-STR or mtDNA filtering, software that computes a “kinship index” for STR profiles skims the database for first-degree relatives (parent–child and sibling relationships) based on the pattern of allele sharing and the frequency of the shared alleles.446
The policy questions are whether exposing relatives to the possibility of being investigated on the basis of genetic leads from their kin is fair and whether the benefits of kinship trawling outweigh the dangers of intrusions from false
444. Jianye Ge & Bruce Budowle, Forensic Investigation Approaches of Searching Relatives in DNA Databases, 66 J. Forensic Sci. 430 (2021), https://doi.org/10.1111/1556-4029.14615 (explaining the advantages and noting the costs of this form of kinship trawling).
445. On this term and possibly clearer alternatives to it, see David H. Kaye, The Genealogy Detectives: A Constitutional Analysis of “Familial Searching,” 50 Am. Crim. L. Rev. 109, 120–21 (2013). “Familial searching” became prominent in the United States with the conviction of the serial killer dubbed the Grim Sleeper, who seemed to take long breaks between murders of young women in South Los Angeles from the mid-1980s until at least 2007. Mike McPhate, Los Angeles Man Is Convicted in ‘Grim Sleeper’ Serial Killer Case, N.Y. Times, May 5, 2016. California authorities located him via the DNA profile of his son, whose profile was in the state database as the result of a 2008 arrest for firearm and drug offenses. Marisa Gerber, The Controversial DNA Search that Helped Nab the ‘Grim Sleeper’ Is Winning over Skeptics, L.A. Times, Oct. 25, 2015. A subsequent and even more highly publicized development—involving trawls of nongovernmental databases for large blocks of SNPs that reveal much more distant relationships—is discussed infra in the section titled “Investigative Genetic Genealogy.” For clarity, the term “familial searching” should be reserved for the trawls, like the one in the Grim Sleeper case, for close relatives in law-enforcement databases.
446. The database profiles with the largest kinship-index values rise to the top. A siblingship index, a paternity index, or any other kinship index is a kind of likelihood ratio (a term explained supra section titled “(1) Definition of the likelihood ratio”). It contrasts the probability of observing the STR types when the profile in the database and the profile of the unknown source of the crime-scene sample have a certain relationship (for example, siblingship) to the probability of observing these types when they are unrelated. Jianye Ge et al., Comparisons of Familial DNA Database Searching Strategies, 56 J. Forensic Sci. 1448, 1448 (2011), https://doi.org/10.1111/j.1556-4029.2011.01867.x; Steven P. Myers et al., Searching for First-Degree Familial Relationships in California’s Offender DNA Database: Validation of a Likelihood Ratio-Based Approach, 5 Forensic Sci. Int’l: Genetics 493 (2011), https://doi.org/10.1016/j.fsigen.2010.10.010.
leads, disruption of family harmony, and exposure of family or individual secrets, such as misattributed paternity.447 The extent to which such concerns could support constitutional challenges to kinship trawling has been debated.448 A few states have banned kinship trawling of their law-enforcement databases, and several jurisdictions subject it to “an approval process, specified and limited offense types, and an affirmative assertion by specific stakeholders that all leads have been exhausted.”449
All-pairs matching within a database to verify estimated random-match probabilities
A third and final use of police intelligence databases has evidentiary implications. The section above titled “Frequencies, Probabilities, and Prejudice” explained the use of population-genetics models to estimate DNA-genotype frequencies. Large databases can be used to check these theoretical computations. Using the New Zealand national database, which consisted of six-locus profiles, for example, researchers compared every profile with every other profile.450 At the time, there were 10,907 profiles in the database. Although this number is not huge, 10,907 profiles can be arranged to form about 59 million distinct pairs.451 Because the theoretical random-match probability was about 1 in 50 million, if all the individuals represented in the database were unrelated, one would expect that an exhaustive comparison of the profiles for these
447. See, e.g., Henry T. Greely et al., Family Ties: The Use of DNA Offender Databases to Catch Offenders’ Kin, 34 J.L. Med. & Ethics 248 (2006), https://doi.org/10.1111/j.1748-720X.2006.00031.x; Erica Haimes, Social and Ethical Issues in the Use of Familiar Searching in Forensic Investigations: Insights from Family and Kinship Studies, 34 J.L. Med. & Ethics 263 (2006), https://doi.org/10.1111/j.1748-720X.2006.00032.x; Erin Murphy, Relative Doubt: Familial Searches of DNA Databases, 109 Mich. L. Rev. 291 (2010); Murphy, supra note 287, at 189–214; Sonia M. Suter, All in the Family: Privacy and DNA Familial Searching, 23 Harv. J.L. & Tech. 309 (2010).
448. Compare Murphy, supra note 447, and Murphy, supra note 287, at 208–11, with Kaye, supra 445.
449. Ge & Budowle, supra note 444, at 432; see also Tepring Piquado et al., Forensic Familial and Moderate Stringency DNA Searches: Policies and Practices in the United States, England, and Wales (2019), https://perma.cc/8HPE-V32Y.
450. Walsh & Buckleton, supra note 439, at 463.
451. For the people represented in the New Zealand database, about 10,907 × 10,907 pairs—such as (1,1), (1,2), (1,3), . . . , (1,10907), (2,1), (2,2), (2,3), . . . , (2,10907), . . . , (10907,1), (10907,2), (10907,3), (10907,10907)—can be formed. This amounts to almost 119 million possible pairs. But there is no point in checking the pairs (1,1), (2,2), . . . (10,907,10,907)—a profile obviously matches itself. Thus, the number of ordered pairs with different individuals is 119 million minus 10,907. Finally, ordered pairs such as (1,5) and (5,1) involve the same two people. Therefore, the number of distinct pairs of people is about half of 119 million—the 59 million figure in the text. More generally, an all-pairs search in a large database of size N involves N(N–1)/2, or about N2/2 comparisons.
59 million pairs would produce only about one match. In fact, the 59 million comparisons revealed 10 matches. What could have caused this excess number of matches? Further investigation showed that eight of the pairs were twins or brothers, the ninth was a duplicate (because one person gave a sample as himself and then again pretending to be someone else), and the tenth was apparently a match between two unrelated people. This exercise thus confirmed the theoretical computation of the random-match probability. On average, the theoretical match probability was about 1/50,000,000, and the rate of matches in the unrelated pairs within the database was 1/59,000,000.
In the United States, defendants have argued that the unintuitively high number of partial matches from internal all-pairs trawls of offender databases demonstrates that the usual estimates of random-match probabilities are incorrect,452 and they have sought discovery of the criminal-offender databases to determine whether the number of matching and partially matching pairs exceeds the predictions made with the population-genetics model.453 An early report about partial matches (at any nine loci out of the 13 CODIS core loci) in the state database in Arizona was said to show extraordinarily large numbers of partial matches (without accounting for the combinatorial explosion in the number of comparisons in an all-pairs database search).454 However, some
452. E.g., People v. Luna, 989 N.E.2d 655, 664–65 (Ill. App. Ct. 2013) (defense expert’s testimony on results of an all-pairs trawl of the Illinois state dataset).
453. United States v. Davis, 602 F. Supp. 2d 658, 681 (D. Md. 2009) (reporting results from an all-pairs search of the Maryland database ordered by a state court); Derr v. State, 73 A.3d 254 (Md. 2013) (defendant who matched at 13 loci was not entitled to have the FBI determine how many pairwise matches there would be at 9 through 13 loci; neither was he entitled to all the profiles in the national database so that his experts could conduct these pairwise comparisons); State v. Dwyer, 985 A.2d 469 (Me. 2009) (defendant was not entitled to require the state to perform an all-pairs search of its database for matches at nine or more loci); Young v. United States, 63 A.3d 1033 (D.C. 2013) (defendant who matched at 13 loci was not entitled to all-pairs searching of the national database); Jason Felch & Maura Dolan, How Reliable Is DNA in Identifying Suspects?, L.A. Times, July 20, 2008.
454. Felch & Dolan, supra note 453; David H. Kaye, Trawling DNA Databases for Partial Matches: What Is the FBI Afraid Of?, 19 Cornell J.L. & Pub. Pol’y 145 (2009); Murphy, supra note 287, at 107–08. As illustrated, supra note 451, an all-pairs search in a large database of size N involves nearly N2/2 comparisons. For example, a database of 6 million samples gives rise to some 18,000,000,000,000 comparisons. Even with no population structure, relatives, and duplicates, and with random-match probabilities in the trillionths, one would expect to find a large number of matches or near-matches. An analogy can be made to the famous “birthday problem.” In its simplest form, the birthday problem assumes that equal numbers of people are born every day of the year. The problem is to determine the minimum number of people in a room such that the odds favor there being at least two of them who were born on the same day of the same month. Focusing solely on the random-match probability of 1/365 for a specified birthday makes it appear that a huge number of people must be in the room for a match to be likely. After all, the chance of a match between two individuals having a given birthday (say, January 1) is (ignoring leap years) a minuscule 1/365 × 1/365 = 1/133,225. But because the matching birthday can be any
scientists question the utility of the investigative databases for population-genetics research.455 They observe that these databases contain an unknown number of relatives, that they might contain duplicates, and that the population in the offender databases is highly structured. These complicating factors would need to be considered in testing for an excess of matches or partial matches. Studies of offender databases in Australia and New Zealand that made adjustments for population structure and close relatives showed substantial agreement between the expected and observed numbers of partial matches, at least up to the nine STR loci used for database profiles in those countries at the time of the studies.456
The existence of large databases also provides a means of estimating a random-match probability without making any modeling assumptions. For the New Zealand study, even ignoring the possibility of relatives and duplicates, there were only 10 matches out of 59 million comparisons. The empirical estimate of the random-match probability is therefore about 1 in 5.9 million. This is about 10 times larger than the theoretical estimate, but still quite small. As this example indicates, crude but simple empirical estimates from all-pairs comparisons in large databases may well produce random-match probabilities that are larger than the theoretical estimates (as expected when full siblings or other close relatives are in the databases), but the estimated probabilities are likely to remain impressively small.
Investigative Genetic Genealogy (IGG)
Until recently, personal knowledge, oral histories, and documents were the only ways to connect extended family members within and across generations. Since
one of the 365 days in the year and because there are N(N–1)/2 ways to have a match, it takes only N = 23 people before it is more likely than not that at least two people share a birthday. The birthday problem thus shows that surprising coincidences commonly occur even in relatively small databases. See, e.g., Persi Diaconis & Frederick Mosteller, Methods for Studying Coincidences, 84 J. Am. Stat. Ass’n 853 (1989).
455. Bruce Budowle et al., Partial Matches in Heterogeneous Offender Databases Do Not Call into Question the Validity of Random Match Probability Calculations, 123 Int’l J. Legal Med. 59 (2009), https://doi.org/10.1007/s00414-008-0239-1.
456. James M. Curran et al., Empirical Support for the Reliability of DNA Evidence Interpretation in Australia and New Zealand, 40 Australian J. Forensic Sci. 99, 102–06 (2008), https://doi.org/10.1080/00450610802172230; Bruce S. Weir, The Rarity of DNA Profiles, 1 Annals Applied Stat. 358 (2007), https://doi.org/10.1214/07-AOAS128; Bruce S. Weir, Matching and Partially-Matching DNA Profiles, 49 J. Forensic Sci. 1009, 1013 (2004); cf. Laurence D. Mueller, Can Simple Population Genetic Models Reconcile Partial Match Frequencies Observed in Large Forensic Databases?, 87 J. Genetics (India) 101 (2008), https://doi.org/10.1007/s12041-008-0016-4 (maintaining that excess partial matches in an Arizona offender database are not easily reconciled with theoretical expectations). This literature is reviewed in Kaye, supra note 454.
2007, direct-to-consumer genetic testing (DTC-GT) has made it possible to find relatives through inherited DNA. Tens of millions of people have undergone such testing for information about their ancestral origin and potential biological relation to other people within the vendors’ databases.457 Law-enforcement officials also have turned to some of these databases to track down the source of crime-scene DNA or to identify human remains indirectly. Like the members of the public interested in finding possible relatives, they try to learn from the companies that assemble these databases if DNA data from potential relatives of the source of the forensic sample seem to be included in the genealogy databases. Constructing parts of the family tree of these relatives has led investigators to the source of the forensic DNA in hundreds of cases.458 This section explains how this adaptation of personal genetic genealogy is conducted, how it differs from familial searching with law-enforcement databases, how it might be regulated, and how it might implicate the Fourth Amendment. We begin with a brief introduction to the use of DTC-GT databases in genealogy research by members of the public.
How does direct-to-consumer genetic genealogy work?
A DTC-GT company supplies paying customers with mail-in spit kits or cheek swabs. Upon receiving the sample, the company uses certain microarrays (the SNP chips described in the section above titled “Sequence-Specific Probes and Microarrays”) to type hundreds of thousands of SNPs. These SNPs are spaced across the 22 autosomal chromosomes, somewhat like individual letters sampled from the volumes of an encyclopedia without the intervening text. Thus, they are not themselves DNA sequences.
Starting in 2009, DTC-GT companies began assembling this information into databases to assist customers seeking to discover possible relatives.459 A DTC-GT customer can have the company trawl the database with specialized software to identify the chromosomal locations and lengths of blocks of SNPs (haploblocks) that match between the customer’s data file and the files
457. James W. Hazel et al., Direct-to-Consumer Genetic Testing: Prospective Users’ Attitudes Toward Information About Ancestry and Biological Relationships, 16 PLoS One e0260340 (2021), https://doi.org/10.1371/journal.pone.0260340. “This genetic genealogy has enabled thousands of individuals who have lost their biological identity through adoption, abandonment, anonymous gamete donation, misattributed parentage, etc., to regain their genetic heritage.” Ellen M. Greytak et al., Genetic Genealogy for Cold Case and Active Investigations, 299 Forensic Sci. Int’l 103, 103 (2019), https://doi.org/10.1016/j.forsciint.2019.03.039.
458. Tracey L. Dowdeswell, Forensic Genetic Genealogy: A Profile of Cases Solved, 58 Forensic Sci. Int’l: Genetics 102679 (2022), https://doi.org/10.1016/j.fsigen.2022.102679.
459. Daniel Kling et al., Investigative Genetic Genealogy: Current Methods, Knowledge and Practice, 52 Forensic Sci. Int’l: Genetics 102474, at 2 (2021), https://doi.org/10.1016/j.fsigen.2021.102474.
in the company’s database.460 Large matching haploblocks—measured in centimorgans461—are believed to be “identical by descent” (IBD) and thus indicative of a common recent ancestor.462 As a rule, the greater the total length of the shared IBD haploblocks, the closer the relationship.463 The total length thus indicates “the probability that the relationship between the unknown individual and the match falls into each degree of relatedness.”464 But different relationships can correspond to the same range of haploblock sharing.465 For example, a total of 100 centimorgans could indicate “anywhere from 5th degree to >8th degree, with 6th degree being most likely.”466 Consequently, the customer may have to explore multiple possibilities.467 Family records, obituaries, and other components of traditional genealogical research will contribute to the proper construction of the family tree.
Someone who wants to maximize the chance of finding relatives can repeat the entire process with another DTC-GT company, but a more efficient strategy is to download the file listing the individual’s SNPs and to submit it to other database companies that are willing to accept data files that they did not
460. See Greytak et al., supra note 457, at 107.
461. On average, a 1 centimorgan (cM) haploblock is about 1 million base pairs long, but a centimorgan is not a fixed physical distance. One cM equates to a 1% probability of recombination within the haploblock when the egg and sperm cells are formed (see supra section titled “Sexual Reproduction and the Genome”). Because recombination occurs more often in certain places along the genome than others, the physical distance depends on the location within the genome. Also, because recombination during the formation of egg cells happens more frequently than in the formation of sperm cells, the physical distance also differs between males and females. The randomness of the points at which “crossing over” of chromosomes occurs during meiosis gives rise to the relationship between total haploblock length and types of kinship. For a more complete explanation, see Greytak et al., supra note 457, at 104–06.
462. Claire L. Glynn, Bridging Disciplines to Form a New One: The Emergence of Forensic Genetic Genealogy, 13 Genes 1381, at 4 (2022), https://doi.org/10.3390/genes13081381; Brenna M. Henn et al., Cryptic Distant Relatives Are Common in Both Isolated and Cosmopolitan Genetic Samples, 7 PLoS One e34267 (2012), https://doi.org/10.1371/journal.pone.0034267 (second to ninth degree cousins); Chad Huff et al., Maximum-Likelihood Estimation of Recent Shared Ancestry (ERSA), 21 Genome Rsch. 768 (2011), https://doi.org/10.1101/gr.11597 (third and even fourth degree cousins). However, “the amount of shared DNA can vary greatly for relatives of the same degree,” and “~10% of third cousins and ~50% of fourth cousins share no detectable IBD segments.” Greytak et al., supra note 457, at 106.
463. Ge & Budowle, supra note 444, at 434.
464. Greytak et al., supra note 457, at 107. A given individual has a first-degree relationship with his or her parents, full siblings, and children; a second-degree relationship with grandparents, aunts and uncles, half siblings, nieces and nephews, and grandchildren; a third-degree relationship with great-grandparents, great-uncles and great-aunts, first cousins, half-nieces and half-nephews, grand-nieces and grand-nephews, and great-grandchildren. For a chart, see id. at 105 (fig. 1).
465. Id. at 106 (tbl. 3) & 107; Glynn, supra note 462, at 5–6.
466. Greytak et al., supra note 457, at 106.
467. Id. at 107. Additional complications can arise from the history of mating in some groups. Id. (noting endogamy and “pedigree collapse”).
produce. Indeed, one genealogy resource, known as GEDmatch,468 does no DNA testing but lets anyone upload a SNP genotype file prepared by any major DTC-GT company. It will then perform an autosomal-haploblock comparison. The individual user receives, at no charge, information on total haploblock sharing with other GEDmatch users and whatever name (which could be an alias) and email address (which could be a one-time-use address) the possible relatives provided. In all these situations, the genealogy enthusiast never sees the raw SNP data file of anyone else.
How does IGG work?
IGG applies the same type of kinship searching to DNA recovered from a crime scene or victim to link an individual to the commission of the crime, or from human remains to identify a missing person.469 Also called forensic investigative genetic genealogy (FIGG), IGG differs from familial searching (see section above titled “Kinship trawling (‘familial searching’)”). Familial searching operates within law-enforcement-managed databases and compares STR profiles derived from legally compelled DNA samples. The grasp of this autosomal STR-based kinship trawling is basically limited to first-degree relatives of convicted offenders or arrestees. IGG is another type of “indirect” or “outer-directed” genetic search for individuals who deposited DNA associated with crimes but whose DNA is not in the accessed database. But IGG uses different DNA variants, different software, and different databases. The genealogy databases are populated with SNP DNA profiles that have been voluntarily made available to commercial database vendors for genealogical purposes. Through individually declared consent and privacy options, users of these databases can specify whether they are willing to accept law-enforcement trawling of their information. Unlike familial searching, IGG can support kinship associations beyond first-degree relatives, occasionally looking as far as ninth-degree relatives. As such, policies and legal conclusions reached for familial searching do not necessarily carry over to IGG.
IGG requires three steps to generate leads for police to consider. To begin with, as with all DNA methods, trace-evidence or victim or missing-person
468. A genetic-technology company, Verogen, specializing in forensic-sequencing methods, acquired GEDmatch in 2019, and a larger biotechnology company, Qiagen, acquired Verogen in 2023. Press Release, QIAGEN Completes Acquisition of Verogen, Strengthening Leadership in Human ID/Forensics with NGS Technologies, Jan. 9, 2023, https://perma.cc/8UCR-AEKV.
469. For a comprehensive and detailed review, see Daniel Kling et al., Investigative Genetic Genealogy: Current Methods, Knowledge and Practice, 52 Forensic Sci. Int’l: Genetics 102474 (2021), https://doi.org/10.1016/j.fsigen.2021.102474.
DNA must be recovered. This DNA normally will have been analyzed to yield an STR profile suitable for comparison with a known suspect’s DNA or for trawling a CODIS database. If conventional STR profiling fails, or if queries within CODIS do not yield any matches or are not feasible because of sample limitations,470 a more laborious genetic genealogy investigation might proceed. For such an investigation, police agencies often turn to commercial forensic-science service providers, although publicly funded forensic-science laboratories are developing these capabilities.471
From the forensic sample to the SNP genotype.
The participating laboratory could analyze the DNA in the forensic sample with a SNP chip, as is done for the personal genealogy samples discussed in the preceding section, but microarrays may be less effective with lower quantity, degraded, or contaminated forensic samples.472 To cope with such samples, massively parallel sequencing methods such as whole-genome or targeted sequencing473 have been used to identify upward of thousands to millions of SNPs and pull out a set that are either in the DTC-GT microarrays or are compatible with the databases.474 With a data file from the forensic DNA, the investigation proceeds in essentially the same manner as the usual personal genetic genealogy search.
470. When the quantity of DNA in the forensic sample is very small, and it is doubtful that STR profiling will succeed, rather than waste the DNA, a SNP profile may be generated without first trying STR profiling. In an urgent case, with enough forensic DNA, simultaneously pursuing both STR and haploblock strategies could be attractive.
471. Press Release, Univ. N. Tex. Health Sci. Cntr. at Fort Worth, CHI Becomes Nation’s First Public Lab to Earn Accreditation to Perform Forensic Genetic Genealogy (Oct. 12, 2023), https://perma.cc/F9PW-U9PQ.
472. Kling et al., supra note 469, § 7.1. But see Greytak et al., supra note 457, at 103 (reporting success with trace-evidence samples of as little as one nanogram of DNA).
473. See supra section titled “Sequencing and SNPs.”
474. Ge & Budowle, supra note 444, at 433; Kling et al., supra note 469, § 7.2. A targeted-sequencing kit that generates a smaller data file of about 10,000 widely spaced SNPs with no known clinically meaningful associations has been developed for use with GEDmatch. June Snedecor et al., Fast and Accurate Kinship Estimation Using Sparse SNPs in Relatively Large Database Searches, 61 Forensic Sci. Int’l: Genetics 102769 (2022), https://doi.org/10.1016/j.fsigen.2022.102769; Jessica Watson et al., Operationalisation of the ForenSeq® Kintelligence Kit for Australian Unidentified and Missing Persons Casework, 68 Forensic Sci. Int’l: Genetics 102972 (2024), https://doi.org/10.1016/j.fsigen.2023.102972; see also Andreas Tillmar et al., The FORCE Panel: An All-in-One SNP Marker Set for Confirming Investigative Genetic Genealogy Leads and for General Forensic Applications, 12 Genes (Basel) 1968 (2021), https://doi.org/10.3390/genes12121968 (targeted sequencing kit said to include “3931 autosomal SNPs for extended kinship analysis” and “designed to exclude clinically relevant markers”).
From the SNP genotype to possible relatives.
The IGG service provider uploads the SNP genotype to the website of a company that maintains a database of SNP profiles for individuals looking for possible relatives (and that is willing to accept data files that it did not generate). Most if not all of these companies have procedures that allow customers to affirmatively opt in or opt out of warrantless forensic IGG trawls.475 The company’s software performs haploblock matching within its database. The matching involves only the length of shared haploblocks;476 there is no need for the analyst to look at which SNPs are present within these or any other haploblocks in the forensic sample.477 The database company then informs the investigators of the names or usernames of individuals with enough shared haploblocks to merit attention as possible relatives. The total extent and pattern of the overlap in the SNP genotypes among the “matches” will be available, along with a way to contact the possible relatives. If their identities can be ascertained, traditional genealogy work begins. A genealogist constructs family trees (typically with hundreds of entries for a single third-cousin-level match), usually by scouring public records, newspapers, social media, and so on.478 The goal is to find a common ancestor for a SNP profile in the genealogy database and the forensic-sample profile. Descendants of that ancestor will include the unknown DNA source, and a combination of the demographic and genetic information may enable a genealogist or crime analyst with genealogy training to whittle down the list of descendants somewhat.479 The police now have their investigative lead. Often, it is followed by surreptitiously collecting DNA from the individuals in question to ascertain if any of them have the same STR profile as that of the forensic DNA.480
Does IGG violate the Fourth Amendment?
A series of Fourth Amendment questions can arise in connection with IGG. At the outset, one might ask whether the steps involved in IGG are a “search or
475. Heather Murphy, What You’re Unwrapping When You Get a DNA Test for Christmas, N.Y. Times, Dec. 22, 2019.
476. See supra section titled “How does direct-to-consumer genetic genealogy work?”
477. Such data normally would exist only in cases in which the SNP profile was derived from more complete genome sequence data.
478. Greytak et al., supra note 460, at 108.
479. Id. at 107–10; Ge & Budowle, supra note 444, at 436 (“Most genealogy investigations focus on third degree or closer relationships because of the substantial burden of searching fourth degree or more matches. An efficient way to improve public record searching is triangulation, which identifies a source by intersecting between two potential families.”).
480. Surreptitious DNA sampling is discussed further in the next two sections. In cases in which an STR profile cannot be obtained (see supra note 470), SNP-identification profiles can be used for the direct identification.
seizure” of “persons, houses, papers [or] effects.”481 If they are, a further question is whether taking any or all of those steps without a warrant would violate the Amendment’s protection against “unreasonable searches and seizures.” And finally, if police apply for a warrant, what would be necessary to establish probable cause?
Are parts of IGG a search or seizure?
As explained in the previous section, IGG begins with the collection of biological material from a crime scene or a victim’s remains and the extraction of DNA. To demonstrate a search or seizure, defendants would have to show either (1) that the forensic-evidence collection and preparation interferes with their right to possess or enjoy their property or personal effects482 or (2) that simply taking possession of the stain, fluid, or cells and extracting DNA reveals information in which they have a reasonable or legitimate expectation of privacy.483 Few, if any, defendants have advanced such claims about biological trace evidence.484
The next step in the process is the extensive SNP analysis of the forensic sample. Is this laboratory analysis of an unknown person’s DNA a search? A number of defendants have challenged the taking—and subsequent STR genotyping—of DNA left behind by suspects in police stations or in public places.485 By and large, courts have been persuaded that STR profiling as
481. U.S. Const. amend. IV.
482. See United States v. Jacobsen, 466 U.S. 109, 113 (1984) (“‘seizure’ of property occurs when there is some meaningful interference with an individual’s possessory interests in that property”). Unlike the collection of DNA directly from the person, acquiring DNA from the crime scene or the remains does not entail a bodily intrusion.
483. Katz v. United States, 389 U.S. 347, 353 (1967); Rakas v. Illinois, 439 U.S. 128, 143 (1978).
484. Cf. State v. Hartman, 534 P.3d 423, 431–32 (Wash. Ct. App. 2023) (rejecting defendant’s argument that under a provision of the state’s constitution guaranteeing that “[n]o person shall be disturbed in his private affairs . . . without authority of law,” he “retained a privacy interest” in the DNA extracted from semen on the body of a 12-year-old girl who was found raped and murdered).
485. E.g., United States v. Hicks, No. 2:18-cr-20406-JTF-tmp, 2020 WL 7704556 (W.D. Tenn. May 27, 2020) (noting cases); State v. Burns, 988 N.W.2d 352 (Iowa 2023); cf. State v. Athan, 158 P.3d 27 (Wash. 2007) (detectives, posing as a fictitious law firm, sent the defendant a letter inviting him to join a fictitious class-action lawsuit, to obtain DNA from the return envelope to compare to DNA from the crime scene). Commentators have questioned the “abandonment” line of cases. E.g., Elizabeth E. Joh, Reclaiming “Abandoned” DNA: The Fourth Amendment and Genetic Privacy, 100 Nw. U. L. Rev. 857–84 (2006); Tracey Maclin, Government Analysis of Shed DNA Is a Search Under the Fourth Amendment, 48 Tex. Tech. L. Rev. 287 (2015); Albert E. Scherr, Genetic Privacy and the Fourth Amendment: Unregulated Surreptitious DNA Harvesting, 47 Ga. L. Rev. 445 (2013). But see Edward J. Imwinkelried & David H. Kaye, DNA Typing: Emerging or Neglected Issues, 76 Wash. L. Rev. 413, 437 (2001); David H. Kaye, Science Fiction and Shed DNA, 101 Nw. U. L. Rev. Colloquy 62 (2006), https://perma.cc/596A-NKH7 (response to Joh).
conducted in the laboratory solely to produce identifying information from “abandoned” biological material is no more invasive of privacy than examining fingerprints from a crime scene.486 Singly, or more likely in combination, however, many of the SNPs in the raw data file could serve as markers or predictors of disease.487 Yet, neither the police nor the laboratories working for them inspect the SNP data files for such markers to make haploblock comparisons.488 Is this fact sufficient to defeat the claim that the very possession of a list of SNPs in a text file interferes with a reasonable expectation of privacy (even before the identity of the individual whose DNA has been analyzed is known)?489 If not, would legally enforceable rules against misuse or unauthorized disclosure create a convincing analogy to photographs, fingerprints, or CODIS STR profiles (assuming that the act of comparing such biometric identifiers is not itself a search)? Or is the creation of the SNP data file to find the source of the as-yet-unidentified DNA more like the actual perusal of the text and image files in smartphones490 or the acquisition of cell-site location information on personal movements over long time periods491—both Fourth Amendment searches?
The next step in IGG is the haploblock-sharing trawl. Is the trawl performed by the company that operates the commercial genealogy database a governmental search492 of the defendant’s person or effects? A series of
486. Opinions zeroing in on this issue include United States v. Davis, 690 F.3d 226, 243–46 (4th Cir. 2012) (separate search); Burns, 988 N.W.2d at 364–65 (no search); Raynor v. State, 99 A.3d 753 (Md. 2014) (no search); Commonwealth v. Arzola, 26 N.E.3d 185, 190–94 (Mass. 2015) (no search). For academic commentary on STR profiling as a search unto itself, see, for example, Kaye, supra note 445; Maclin, supra note 485; Murphy, supra note 446.
487. To reduce this risk, sequencing kits that generate smaller data files of widely spaced SNPs with no known clinically meaningful associations have been developed. See supra note 474.
488. Ellen M. Greytak et al., Privacy and Genetic Genealogy Data, 361 Science 857 (2018), https://doi.org/10.1126/science.aav0330; Greytak et al., supra note 457, at 106–07 (“Additionally, no sensitive genetic information is disclosed to law enforcement during a genetic genealogy search, as the raw genetic data from GEDmatch users is not accessible . . . GEDmatch simply performs comparisons among samples, returning the lengths and chromosomal locations of shared DNA segments, which are used to determine the approximate relationship between individuals.”).
489. Initially, the investigators’ SNP data file does not pertain to any known individual, but if IGG is successful and the source of the trace-evidence sample is located, then the data will pertain to that person.
490. Riley v. California, 573 U.S. 373 (2014) (viewing data stored on a cellphone is a search that does not fall within the search-incident-to-arrest exception to the warrant-and-probable-cause requirement).
491. Carpenter v. United States, 585 U.S. 296 (2018).
492. On the question of whether ordinary, non-kinship trawls in law-enforcement STR databases are Fourth Amendment “searches,” see Boroian v. Mueller, 616 F.3d 60 (1st Cir. 2010) (retrawling after a sentence is served is not a search); Johnson v. Quander, 440 F.3d 489, 498–99 (D.C. Cir. 2006) (retrawling after the period of probation is completed is not a search); David H. Kaye, DNA Database Trawls and the Definition of a Search in Boroian v. Mueller, 97 Va. L. Rev. in Brief 41 (2011);
subquestions may need to be considered. Did the company knowingly and voluntarily trawl the database to assist the investigation?493 Or did the company not intend to make its services available to law enforcement,494 but police submitted SNP data files anyway, pretending to be an ordinary subscriber? Would a customer, having chosen to expose his or her SNP data to trawling on behalf of anyone in the general public, retain a reasonable expectation of privacy in that data?495 And, even if the trawl were considered a search of the customer’s data, could a defendant suppress the evidence on the theory that the
Catherine W. Kimel, Note, DNA Profiles, Computer Searches, and the Fourth Amendment, 62 Duke L.J. 933 (2013).
493. FamilyTreeDNA and GEDmatch require law-enforcement agencies to identify themselves as such when submitting a SNP-genotype file. FamilyTreeDNA Law Enforcement Guide, https://perma.cc/PHR5-2B59, last visited, Dec. 16, 2023; GEDmatch, Terms of Service and Privacy Policy, Dec. 30, 2021, https://perma.cc/FJ5G-AL6F.
494. E.g., MyHeritage—Terms and Conditions (June 11, 2023), https://perma.cc/DJP9-D7UZ (“using the DNA Services for law enforcement purposes, forensic examinations, criminal investigations, ‘cold case’ investigations, identification of unknown deceased people, location of relatives of deceased people using cadaver DNA, and/or all similar purposes, is strictly prohibited, unless a court order is obtained”); GEDmatch, Terms of Service and Privacy Policy (Dec. 30, 2021), https://perma.cc/LF67-BBKD (if you “[o]pt-out . . . [y]our kit WILL NOT be compared with kits submitted by law enforcement to identify perpetrators of violent crimes”) (emphasis in original).
495. It could be argued that just because DNA samples are sent to DTC-GT companies for SNP analysis, or because the SNP data are made available to the public for genetic-genealogy trawls, no cognizable search occurs when the government also submits a data file for trawling and receives the results. Cf., e.g., United States v. Miller, 425 U.S. 435, 443 (1976) (“The [customer who maintains a bank account] takes the risk, in revealing his affairs to another, that the information will be conveyed by that person to the Government. . . . [T]he Fourth Amendment does not prohibit the obtaining of information revealed to a third party and conveyed by him to Government authorities, even if the information is revealed on the assumption that it will be used only for a limited purpose and the confidence placed in the third party will not be betrayed.”). In Carpenter v. United States, 585 U.S. 296 (2018), however, the Court clarified that this “third-party doctrine” depends on the nature of the information compiled by or disclosed to third parties. It held that extensive data on the locations of a person’s cell phone relative to cell towers reveals too much about a person’s private life to be accessible to police at almost no cost just because the person’s movements “might be pertinent to an ongoing investigation.” Id. at 317. Carpenter opens the door for subscribers to claim that the genealogy database trawl is a Fourth Amendment “search” of their data. They can argue that exposing them to the risk of being placed on a family tree known to the government is no less invasive of a legitimate privacy interest than a cellphone customer “effectively [being] tailed every moment of every day for five years.” Id. at 312. But there is public and scholarly disagreement on the strength of the interests of the subscribers to genetic genealogy services in the secrecy of their biological affiliations and their SNPs. Compare James W. Hazel & Christopher Slobogin, “A World of Difference”? Law Enforcement, Genetic Data, and the Fourth Amendment, 70 Duke L.J. 705 (2021); Ayesha K. Rasheed, Personal Genetic Testing and the Fourth Amendment, 2020 U. Ill. L. Rev. 1249; and Natalie Ram, Genetic Privacy After Carpenter, 105 Va. L. Rev. 1357 (2019); with Teneille R. Brown, Why We Fear Genetic Informants: Using Genetic Genealogy To Catch Serial Killers, 21 Colum. Sci. & Tech. L. Rev. 114 (2019); cf. Paul Ohm, The Many Revolutions of Carpenter, 32 Harv. J.L. & Tech. 357, 385 (2019) (concluding that even for a database of whole-genome
use of the customer’s data unreasonably invaded the customer’s Fourth Amendment interests?496
Warrants and probable cause?
If some or all of these steps in IGG are individually or collectively a Fourth Amendment search of the defendant’s person, papers, or effects, two broad questions remain: first, whether such searches are constitutionally unreasonable in the absence of a warrant based on probable cause; and, second, if a warrant is sought, what must be shown to establish probable cause. With regard to reasonableness, a few Supreme Court opinions suggest that “our general Fourth Amendment approach [entails] ‘examining the totality of the circumstances. . . .’”497 In IGG cases, parties might argue about the relevance and impact of many circumstances. For example, did anyone actually scrutinize the defendant’s SNP profile for medically or socially sensitive information? Are there statutory, administrative, or technical constraints against using the SNP data for such purposes?498 Is the system designed to avoid unnecessary disclosures of family information, such as misattributed paternity? Is the prospect that law-enforcement authorities will initiate trawls of the database known to the subscribers?499 These privacy-related factors might affect the magnitude of the individual interests as compared to government’s law-enforcement interest.
But directly balancing the two sets of interests to determine the reasonableness of a Fourth Amendment search or seizure is controversial. More often, the Supreme Court has admonished that “searches conducted outside the judicial
sequences “[i]t seems unlikely that a court would require a warrant for DNA evidence held by a private third party based on a straight application of the Carpenter factors”).
496. The defendant would have to overcome the doctrine that an impermissible intrusion on the security or privacy of another person’s papers or effects does not entitle the defendant to suppress the evidence. See, e.g., Rakas v. Illinois, 439 U.S. 128, 133–34 (1978) (“A person who is aggrieved by an illegal search and seizure only through the introduction of damaging evidence secured by a search of a third person’s premises or property has not had any of his Fourth Amendment rights infringed.”); Minnesota v. Carter, 525 U.S. 83, 91 (1998) (a temporary guest in an apartment does not have standing to challenge the reasonableness of a search of the apartment).
497. United States v. Knights, 534 U.S. 112, 118 (2001). With CODIS STR databases, lower courts using this “totality” approach typically balanced the government’s interests in building and operating these intelligence-gathering databases against the individual’s legitimate interests in being free from bodily intrusion and maintaining the confidentiality of the STR data and samples. See supra section titled “Law-enforcement databases and databanks.”
498. Cf. Maryland v. King, 569 U.S. 435 (2013) (DNA sampling and CODIS STR profiling of arrestees); Whalen v. Roe, 429 U.S. 589 (1977) (law requiring that a copy of prescriptions for the most dangerous legitimate drugs be sent to the state for computer scanning, storage, and access under secure conditions was a reasonable exercise of state police power).
499. See supra section titled “From the SNP genotype to possible relatives” (on policies allowing customers to opt in or opt out of exposure to database trawls for law-enforcement purposes).
process, without prior approval by judge or magistrate, are per se unreasonable under the Fourth Amendment—subject only to a few specifically established and well-delineated exceptions.”500 Under this “warrant preference rule,” balancing to determine the reasonableness of a type of search that dispenses with warrants or probable cause still might occur, but only at the level of delineating a new categorical exception to these procedures.501 For a warrant to issue, a magistrate would need to verify that the police have probable cause. But probable cause to believe what? The Supreme Court has said that probable cause to arrest or search a person exists when the totality of the circumstances indicates “reasonable ground for belief of guilt,” and “the belief of guilt [is] particularized with respect to the person to be searched or seized.”502 Genealogy database trawls are not undertaken because of a belief about a particular person. They are initiated in the hope that some individuals unknown to the police have their SNP genotypes in the database and that the company’s haploblock-matching software will flag them as possible relatives. Should the probable-cause finding turn on whether the size and likely composition of a company’s database indicates a reasonable probability of a useful match occurring in that particular database? Why not all databases combined, since the trawls could be performed in all of them? Or are such warrants too redolent of “the reviled ‘general warrants’ and ‘writs of assistance’ of the colonial era, which allowed British officers to rummage through homes in an unrestrained search for evidence of criminal activity”?503 The nature of probable cause in the context of IGG may need to be explored.
How should IGG be regulated?
After IGG burst into the public consciousness with the arrest in 2018 of a former police officer found to be the elusive Golden State Killer,504 DTC-GT companies scrambled to develop clearer or different policies,505 and observers advocated
500. Katz v. United States, 389 U.S. 347, 357 (1967) (notes omitted).
501. David H. Kaye, A Fourth Amendment Theory for Arrestee DNA and Other Biometric Databases, 15 U. Pa. J. Const. L. 1095, 1139–40 (2013) (proposing a “biometric exception” that would permit warrantless searches of fingerprint, photograph, and, arguably, CODIS STR databases). For efforts to explain the rationale for the balancing for DNA sampling that the Court performed in Maryland v. King, see, for example, Kaye, 104 J. Crim. L. & Criminology 535, supra note 431; Maclin, supra note 431.
502. Maryland v. Pringle, 540 U.S. 366, 371 (2003) (internal quotation marks and citations omitted).
503. Riley v. California, 573 U.S. 373 (2014).
504. See, e.g., Paige St. John, The Untold Story of How the Golden State Killer Was Found: A Covert Operation and Private DNA, L.A. Times, Dec. 8, 2020.
505. Glynn, supra note 462, at 10; Murphy, supra note 475.
for external rules and oversight of when and how police access the databases.506 A year and a half later, the Department of Justice settled on an “interim policy” for its agencies, employees, and contractors.507 This policy
- Mostly limits the use of IGG to unsolved homicides and sex crimes.508
- Restricts investigators to “only those GG services that provide explicit notice to their service users and the public that law enforcement may use their service sites.”509
- Requires that “biological samples and [IGG] profiles [be used] only for law enforcement identification purposes”510 and not “to determine the sample donor’s genetic predisposition for disease or any other medical condition or psychological trait.”511
- Establishes procedural constraints on surreptitious collection of DNA from certain individuals who are not targets of the investigation.512
- Requires that all forensic samples first be tested to generate an STR profile prior to performing IGG, a practice that is not feasible with low input, degraded, and rootless hair samples.
506. Sara H. Katsanis, Pedigrees and Perpetrators: Uses of DNA and Genealogy in Forensic Investigations, 21 Ann. Rev. Genomics & Hum. Genetics 535 (2020), https://doi.org/10.1146/annurev-genom-111819-084213; Erin Murphy, Law and Policy Oversight of Familial Searches in Recreational Genealogy Databases, 292 Forensic Sci. Int’l e5–e9 (2018), https://doi.org/10.1016/j.forsciint.2018.08.027; Paige St. John, DNA Genealogical Databases Are a Gold Mine for Police, But with Few Rules and Little Transparency, L.A. Times, Nov. 24, 2019.
507. U.S. Dep’t of Just., Interim Policy Forensic Genetic Genealogical DNA Analysis and Searching, Nov. 1, 2019, https://perma.cc/D6ZF-D6SW. The policy also applies to “any federal agency or any unit of state, local, or tribal government that receives grant award funding from the Department that is used to conduct FGG/FGGS.” Id. at 2. However, the policy “does not . . . limit the prerogatives, choices, or decisions available to, or made by, the Department in its discretion.” Id. at 1 n.1.
508. These include missing-person cases where the unidentified human remains are reasonably believed to be those of a suspected homicide victim. Id. at 4. However, IGG can be used for other crimes when (1) they are “serious” and the database vendor authorizes “investigative use of its service by law enforcement,” id. at 4 n.15, or (2) “the circumstances surrounding the criminal act(s) present a substantial and ongoing threat to public safety or national security.” Id. at 5. Reasonable investigative leads must have been pursued, and a CODIS search must have failed. Id.
509. Id. at 6.
510. Id.
511. Id. at 7.
512. The protected individuals are non-suspects “who may have a closer kinship relationship to the donor of the forensic sample than the associated [genetic genealogy] service user” and who have come to light as a result of the genealogical research. Id. at 6. In such cases, investigators either must obtain their informed consent or have “reasonable grounds to believe that this request would compromise the integrity of the investigation” and consult prosecutors before covertly collecting their samples. Id. In addition, investigators must obtain a search warrant for “a vendor laboratory” to perform SNP typing on the sample. Id.
- Requires that only STR profiles be used for confirmatory identity verification against a sample collected from the putative perpetrator or missing person, when SNP-based statistics might more easily be generated with certain samples.
- Creates other constraints and procedural requirements as well.
In sum, the interim policy stakes out a middle ground between unfettered IGG and a total ban. A few states have enacted more restrictive statutes that include judicial oversight.513 Both the Justice Department policy and the statutes have their critics.514
Inferring Traits
Kinship searching (see section titled “Kinship trawling (‘familial searching’) above)” is one technological response to unsuccessful database searches. Two additional techniques are available: biogeographic ancestry testing and forensic DNA phenotyping. Their purpose is only to focus an investigation by supplying police with some visible or other characteristics that the source of the trace evidence might have. They are not routinely used and are not designed to produce evidence for trial.515 If a suspect is located with the help of these probabilistic clues, STR profiling (or typing with other loci suitable for individual identification) would be undertaken before trial.516
513. E.g., Utah Code § 53-10-403.7 (2023); Natalie Ram et al., Regulating Forensic Genetic Genealogy: Maryland’s New Law Provides a Model for Others, 373 Science 1444 (2021), https://doi.org/10.1126/science.abj5724.
514. See Virginia Hughes, Two New Laws Restrict Police Use of DNA Search Method: Maryland and Montana Have Passed the Nation’s First Laws Limiting Forensic Genealogy, the Method that Found the Golden State Killer, N.Y. Times, May 31, 2021; Laura Geary, A Critical Eye Toward Commercial DNA Database Criminal Procedures, U. Chi. L. Rev. Online, July 8, 2022, https://perma.cc/TG3D-U2GZ (questioning the statutes); Christi Guerrini et al., State Genetic Privacy Statutes: Good Intentions, Unintended Consequences?, Bill of Health (Aug. 10, 2023), https://perma.cc/2VGQ-S4L7 (pointing out ambiguities and anomalies); cf. Brown, supra note 495 (critiquing some of the concerns about IGG); Christi J. Guerrini et al., Four Misconceptions About Investigative Genetic Genealogy, 8 J. L. & Biosciences 1 (2021), https://doi.org/10.1093/jlb/lsab001 (urging more nuanced analysis of the ethical and practical issues).
515. But see Jonathan Foox et al., Epigenetic Forensics for Suspect Identification and Age Prediction, 1 Forensic Genomics 83 (2021), https://doi.org/10.1089/forensic.2021.0005 (experiment with DNA-based age estimation offered to rebut offender’s claim that an old DNA sample from a previous case was planted in the newer case for which post-conviction relief was denied).
516. Indeed, police have used the DNA-based information in conducting “mass screens” or “DNA dragnets” of legally consenting residents of a geographic region where serial rapes, murders, or bombings have occurred. E.g., Monroe v. City of Charlottesville, 579 F.3d 380 (4th Cir. 2009). There is a risk that a screening will be restricted on the basis of race or ethnicity to the wrong group. But DNA predictions also could prompt police to change a screen that otherwise would be
Biogeographic ancestry testing (BGAT)
Some DNA sequences are concentrated in some parts of the world. As such, a well-chosen set of loci can be used to infer the geographic area in which an individual’s biological ancestors lived. The specificity and accuracy of these inferences depends on several factors. What is the size and scale of the population-genetics databases used to associate genotypes with geographic regions? How clearly do the alleles in the panel of loci cluster according to geography (or self-declared ancestry, if that is used)? How effective is the statistical method for probabilistically mapping genotypes to geography?
These matters are all subjects of ongoing research. As potential ancestry-informative markers (AIMs), the STR loci used for individual identification are far from ideal. They vary greatly among individuals—every population has many alleles at each locus—but it is roughly the same “many” in most populations. For ancestry assignment, various panels of SNPs that are more strongly correlated to geography have been published.517 Which ancestry-informative SNPs are present in a trace-evidence sample can be determined with most of the methods described above in the section titled “What Are DNA Polymorphisms and How Are They Detected?”518 Whereas clinical and anthropological research studies often use thousands of SNPs, the panels developed for biological trace evidence, which frequently involves low quantity or degraded DNA, consist of smaller sets of AIMs.519
incorrectly concentrated on one group. Imwinkelried & Kaye, supra note 485, at 451. On the equal-protection ramifications of racially focused investigations resulting from eyewitness statements, see Monroe, supra; Brown v. City of Oneonta, 221 F.3d 329 (2d Cir. 2000); Imwinkelried & Kaye, supra note 485, at 446–51.
517. For a review, see Ozlem Bulbul & Kenneth K. Kidd, Forensic Ancestry Inference: Data Requirements, Analysis Methods, and Interpretation of Results, in Forensic DNA Analysis: Technological Development and Innovative Applications 225, 227–29 (Elena Pilli & Andrea Berti eds., 2021).
518. Id. at 229–31; Elena Pilli et al., Biogeographical Ancestry, Variable Selection, and PLS-DA Method: A New Panel to Assess Ancestry in Forensic Samples via MPS Technology, 62 Forensic Sci. Int’l: Genetics 102806 (2023), https://doi.org/10.1016/j.fsigen.2022.102806.
519. Peter Resutik et al., Comparative Evaluation of the MAPlex, Precision ID Ancestry Panel, and VISAGE Basic Tool for Biogeographical Ancestry Inference, 64 Forensic Sci. Int’l: Genetics 102850 (2023), https://doi.org/10.1016/j.fsigen.2023.102850 (“More than twenty forensic panels designed to distinguish continental ancestry have been proposed by the forensic community over the past two decades. These generally comprise less than 200 AIMs, mainly consisting of autosomal SNPs, but also including other types of markers such as microhaplotypes (MHs) and insertion-deletion polymorphisms (INDELs). In addition, the inclusion of uniparental markers (Y chromosomal SNPs and mtDNA) in the same panel or as a complementary approach has been advocated as it enables ascertaining information on the maternal and paternal lineages, which may help resolve ancestry of recently-admixed individuals.”) (references omitted).
The population-reference databases for developing the ancestry-informative SNPs for crime-scene samples are public research databases.520 Their limited coverage of populations around the world constrains the inferred origins to very large areas. Basically, forensic BGAT is most reliable for mapping a sample to a major continental group such as African, Western Eurasian, East and South Asian, or Native American. Because significant numbers of SNPs are employed as markers, established multivariate statistical methods are used to make the classifications.521 Studies of the accuracy of classifications from different providers continue to appear.522
Geneticists and anthropologists typically caution that BGA should not be confused with ethnicity and race. Ethnicity reflects a person’s social and cultural background. Obviously, these are not determined by DNA. However, a person’s ethnicity may be strongly associated with BGA. As a biological classification, the term “race” is considered inappropriate, but self-reported race also is correlated with BGA.523
Forensic DNA phenotyping (FDP)
A phenotype is an observable or testable trait—skin color, height, blood sugar level, diseases, physical activity level, etc. Phenotypes result from gene activity and environmental factors. Forensic DNA phenotyping refers to inferring traits from the DNA in a trace-evidence sample to assist in locating the source. As such, it might be clearer to call it DNA trace-evidence phenotyping. Regardless of nomenclature, researchers and practitioners have focused on deducing
520. The 1000 Genomes Project, Human Genome Diversity Project-Center d’Étude du Polymorphisme Humain (HGDP–CEPH), and HapMap Project databases are commonly used. A short description of their coverage, along with that of the ALFRED database of frequency of polymorphisms in human populations, is at Bulbul & Kidd, supra note 517, at 231–32.
521. Id. at 232–37. Machine-learning methods also are being studied. Eugenio Alladio et al., Multivariate Statistical Approach and Machine Learning for the Evaluation of Biogeographical Ancestry Inference in the Forensic Field, 12 Sci. Reports 8974 (2022), https://doi.org/10.1038/s41598-022-12903-0.
522. Muna Al-Asfi et al., Assessment of the Precision ID Ancestry Panel, 132 Int’l J. Legal Med. 1581 (2018), https://doi.org/10.1007/s00414-018-1785-9; Lauren Atwood et al., From Identification to Intelligence: An Assessment of the Suitability of Forensic DNA Phenotyping Service Providers for Use in Australian Law Enforcement Casework, 11 Frontiers in Genetics 568701 (2021), https://doi.org/10.3389/fgene.2020.568701; Resutik et al., supra note 519; Watson et al., supra note 487.
523. For discussion of populations and race in forensic genetics, see, for example, Jobling, supra note 183; Mark Shriver et al., Getting the Science and the Ethics Right in Forensic Genetics, 37 Nature Genetics 449 (2005), https://doi.org/10.1038/ng0505-449; supra section titled “Could an Unrelated Person Be the Source?”; cf. Alyna T. Khan et al., Race, Ethnicity, and Genetics Working Group, Recommendations on the Use and Reporting of Race, Ethnicity, and Ancestry in Genetic Research: Experiences from the NHLBI TOPMed Program, 2 Cell Genomics 100155 (2022), https://doi.org/10.1016/j.xgen.2022.100155.
externally visible characteristics of the kind that might appear in an eyewitness’s description.524
One such characteristic (or, more precisely, a state that is associated with many aspects of physical appearance) is biological sex. A locus for sex typing has been part of standard STR profiling for over 25 years.525 Eye color is harder to infer for all people because many genes are involved, but only six are currently used in FDP tests. Some classifications (such as brown eye color versus blue) often can be made,526 and, when they are possible, they seem to be generally accurate.527 Classifications of hair and skin pigmentation also are included in some FDP-typing systems whose validity has been studied in a number of populations.528 Predicting other externally visible traits such as height is even harder because these quantitative traits are determined by large numbers of genes, many of which remain unknown. These traits are also influenced by environmental factors such as nutrition, which cannot be predicted from DNA. Although some police agencies have turned to bioinformatics companies that construct pictures of faces from DNA, this procedure has been criticized as unvalidated, not peer reviewed, and beyond existing knowledge.529
In addition to FDP for characteristics of eyes, hair, and skin, estimation of chronological age is possible.530 Mitochondrial DNA changes with age (and not
524. Scientific literature on FDP is referenced in John M. Butler, Recent Advances in Forensic Biology and Forensic DNA Typing: INTERPOL Review 2019–2022, 6 Forensic Sci. Int’l: Synergy 100311 (2023), https://doi.org/10.1016/j.fsisyn.2022.100311.
525. See supra note 48.
526. Eye color depends on how much of a dark brownish-black pigment called eumelanin is in the iris. Eumelanin absorbs light, so individuals who have a lot of eumelanin in their irises have eye colors that appear dark, such as brown. Several genes responsible for controlling eumelanin production in the eye are known. One of the most important (OCA2) is located on chromosome 15. Individuals with two copies of the G nucleotide at a locus near OCA2 produce less eumelanin and are more likely to have blue eyes. Individuals with one or two copies of an A nucleotide at the same SNP locus produce more eumelanin and are more likely to have brown eyes. Intermediate eye colors such as green and hazel are still very hard to predict.
527. E.g., Ersilia Paparazzo et al., A New Approach to Broaden the Range of Eye Colour Identifiable by IrisPlex in DNA Phenotyping, 12 Sci. Reports 12803 (2022), https://doi.org/10.1038/s41598-022-17208-w (describing the system as “well validated”).
528. See European Forensic Genetics Network of Excellence, Making Sense of Forensic Genetics 31 (2017), https://perma.cc/4RSH-5EQM (“Currently, eye colour, hair colour and skin colour can be predicted reliably and with practically useful accuracy from crime scene DNA, but not yet any other externally visible characteristic.”) (quoting Manfred Kayser); cf. Matteo Fabbri et al., Application of Forensic DNA Phenotyping for Prediction of Eye, Hair and Skin Colour in Highly Decomposed Bodies, 11 Healthcare 647 (2023), https://doi.org10.3390/healthcare11050647.
529. Id.; Jobling, supra note 183.
530. Gerontology researchers often distinguish between chronological and biological age (also known as physiological or functional age). Biological aging occurs as damage to various cells and tissues in the body accumulates. Biological age reflects chronological age, genetics (for example, how quickly antioxidant defenses kick in), lifestyle, nutrition, and diseases and other
for the better),531 but the association of these modifications with age is weak. In nuclear DNA, sequences near the ends of the chromosomes shorten with every cell division. Estimates based on this effect are not very precise (typical errors are around ±20 years, perhaps less when using certain blood cells). Better results come from changes to DNA that do not alter the sequence of nucleotides.532 Such “epigenetic” changes occur in connection with gene expression. Messenger RNA (mRNA) participates in this gene expression, and other molecules attach to the DNA in the process that turns gene expression on and off.533 In particular, a methyl group (one carbon atom and three hydrogen atoms) on top of a gene turns it off (or sometimes on). The extent and pattern of this methylation across the genome changes as an individual ages. This phenomenon enables DNA methylation and mRNA to serve as age markers. Analytical procedures for markers in large trace-evidence samples seem to give estimated ages that usually are accurate within about ±3 or 4 years; massively parallel sequencing for methylation in samples containing smaller amounts of DNA is in development534 and has been used in one prominent criminal case.535
FDP has prompted criticism from some legal commentators and bioethicists.536 Proponents of the methods maintain that FDP merely reveals information for which a person leaving a trace-evidence sample could have no plausible expectation of privacy, since visible traits are exposed to the public anyway. Beyond phenotypes like pigmentation, “lifestyle factors, such as smoking, alcohol intake, drug abuse, diet, physical exercise and educational attainment have been reported to impact our epigenome” and to merit research.537 But some commentators regard “lifestyle and environmental characteristics” as included in “the right to privacy.”538 Other writers note that “if FDP techniques
conditions. Biological age is more closely connected to appearance, but chronological age can be more directly useful in investigations.
531. A higher load of point heteroplasmies and an increasing number of large-scale deletions occur.
532. Vidaki & Kayser, supra note 398.
533. See supra section titled “Genes and Gene Products.”
534. Walther Parson, Age Estimation with DNA: From Forensic DNA Fingerprinting to Forensic (Epi)Genomics: A Mini-Review, 64 Gerontology 326 (2018), https://doi.org/10.1159/000486239; Vidaki & Kayser, supra note 398.
535. State v. Avery, 970 N.W.2d 564 (Wis. Ct. App. 2021) (table); Foox et al., supra note 515.
536. Gabrielle Samuel & Barbara Prainsack, Societal, Ethical, and Regulatory Dimensions of Forensic DNA Phenotyping (2019), https://perma.cc/XT53-H35S (surveying literature and summarizing interviews); Pilar N. Ossorio, About Face: Forensic Genetic Testing for Race and Visible Traits, 34 J. L., Med. & Ethics 277 (2006), https://doi.org/10.1111/j.1748-720X.2006.00033.x; Victor Toom et al., Approaching ethical, legal and social issues of emerging forensic DNA phenotyping (FDP) technologies comprehensively, 22 Forensic Sci. Int’l: Genetics e1 (2016), https://doi.org/10.1016/j.fsigen.2016.01.010.
537. Vidaki & Kayser, supra note 398.
538. Matthias Wienroth et al., Ethics as Lived Practice. Anticipatory Capacity and Ethical Decision-Making in Forensic Genetics, 12 Genes 1868 (2021), https://doi.org/10.3390/genes12121868.
were extended to [certain] non-visible characteristics, such as genetic risk of disease, they could reveal personally sensitive, private information, to whoever is doing the testing—which could be of interest to medical insurance companies or certain employers.”539 In addition, there are questions of premature use of methods that have not attained adequate validation and precision, and of effectively communicating uncertainties. Also, there is concern that police investigations informed by surmises about skin color will “be seen as reinforcing racial stereotyping and perceptions of unequal treatment amongst minority communities.”540
Whether trace-evidence samples can be used for phenotyping has not been litigated. For samples collected for storage in law-enforcement databanks, most legislation establishing and funding law-enforcement DNA databases simply uses the broad term “identification” to denote the purpose behind collecting and storing samples, presumably from convicted offenders or arrestees rather than from the trace evidence. However, a few states exclude testing these samples for “physical characteristics, traits or predispositions for disease.”541
539. European Forensic Genetics Network of Excellence, supra note 528. An outpouring of legislation forbids the use of genetic-test results in health insurance and employment. See generally Jean-Christophe Bélisle-Pipon et al., Genetic Testing Insurance Discrimination and Medical Research: What the United States Can Learn from Peer Countries, 25 Nature Med. 1198 (2019), https://doi.org/10.1038/s41591-019-0534-z; Mark A. Rothstein, Time to End the Use of Genetic Test Results in Life Insurance Underwriting, 46 J.L., Med. & Ethics 794 (2018), https://doi.org/10.1177/1073110518804243 (“By the end of the [1990s], 48 states had enacted laws prohibiting genetic discrimination in health insurance and 35 states prohibited genetic discrimination in employment. In 2008, Congress enacted the Genetic Information Nondiscrimination Act (GINA), which outlawed discrimination based on genetic information in health insurance and employment.”) (notes omitted).
540. Weinroth et al., supra note 538, at 32 (quoting Robin Williams).
541. Indiana Code § 10-13-6-16 (“The information contained in the Indiana DNA data base may not be collected or stored to obtain information about human physical traits or predisposition for disease.”); Wyo. Stat. § 7-19-404(c) (2022) (“Only DNA records which directly relate to the identification characteristics of individuals shall be collected and stored in the state DNA database. The information contained in the state DNA database shall not be collected or stored for the purpose of obtaining information about physical characteristics, traits or predisposition for disease.”); cf. R.I. Gen. L. § 12–1.5-10(5) (2022) (“DNA samples and DNA records collected under this chapter shall never be used under the provisions of this chapter for the purpose of obtaining information about physical characteristics, traits or predispositions for disease.”). On the desirability of limiting the loci that may be typed from offender samples in law-enforcement databanks, compare Mark A. Rothstein & Sandra Carnahan, Legal and Policy Issues in Expanding the Scope of Law Enforcement DNA Data Banks, 67 Brook. L. Rev. 127 (2001), with David H. Kaye, Two Fallacies about DNA Data Banks for Law Enforcement, 67 Brook. L. Rev. 179 (2001). A few European countries have laws that affect phenotyping with trace-evidence samples. Henrik Westermark et al., Swiss Inst. Comparative L., The Regulation of the Use of DNA in Law Enforcement, Aug. 28, 2020, at 10, https://perma.cc/B3K6-DK3T.
Glossary of Terms
adenine (A). One of the four bases, or nucleotides, that make up the DNA molecule. Adenine binds only to thymine. See nucleotide.
affinal method. A method for computing the single-locus profile probabilities for a theoretical subpopulation by adjusting the single-locus profile probability, calculated with the product rule from the mixed population database, by the amount of heterogeneity across subpopulations. The model is appropriate even if there is no database available for a particular subpopulation, and the formula always gives more conservative probabilities than the product rule applied to the same database.
allele. In classical genetics, an allele is one of several alternative forms of a gene. A biallelic gene has two variants; others have more. Alleles are inherited separately from each parent, and for a given gene an individual may have two different alleles (heterozygosity) or the same allele (homozygosity). In DNA analysis, the term is applied to any DNA region (even if it is not a gene) used for analysis.
allelic ladder. A mixture of all the common alleles at a given locus. Periodically producing electropherograms of the allelic ladder aids in designating the alleles detected in an unknown sample. The positions of the peaks for the unknown can be compared to the positions in a ladder electropherogram produced near the time when the unknown was analyzed. Peaks that do not match up with the ladder require further analysis.
amplification. Increasing the number of copies of a DNA region, usually by PCR.
amplified fragment length polymorphism (AMP-FLP). A DNA identification technique that uses PCR-amplified DNA fragments of varying lengths. The DS180 locus is a VNTR whose alleles can be detected with this technique.
ancestry informative markers (AIMs). Polymorphisms for a particular DNA sequence that appear in substantially different frequencies among populations from different geographical regions of the world. AIMs are used to infer the geographical origins of the ancestors of an individual, typically by continent or subcontinent. Panels of SNPs are usually used, as they vary more in their frequency across major populations than do the STRs used in individual identification.
antibody. A protein (immunoglobulin) molecule, produced by the immune system, that recognizes a particular foreign antigen and binds to it; if the antigen is on the surface of a cell, this binding leads to cell aggregation and subsequent destruction.
antigen. A molecule (typically found on the surface of a cell) whose shape triggers the production of antibodies that will bind to the antigen.
autoradiograph (autoradiogram, autorad). In RFLP analysis, the X-ray film (or print) showing the positions of radioactively marked fragments (bands) of DNA, indicating how far these fragments have migrated, and hence their molecular weights.
autosome. A chromosome other than the X and Y sex chromosomes.
band. See autoradiograph.
band shift. Movement of DNA fragments in one lane of a gel at a different rate than fragments of an identical length in another lane, resulting in the same pattern “shifted” up or down relative to the comparison lane. Band shift does not necessarily occur at the same rate in all portions of the gel.
base pair (bp). Two complementary nucleotides bonded together at the matching bases (A and T or C and G) along the double helix “backbone” of the DNA molecule. The length of a DNA fragment often is measured in numbers of base pairs (1 kilobase (kb) = 1,000 bp); base-pair numbers also are used to describe the location of an allele on the DNA strand.
Bayes’ factor. A ratio that indicates how much the data change a person’s prior odds according to Bayes’ rule. In simple situations, the Bayes’ factor has the same value as the likelihood ratio.
Bayes’ rule. A formula that relates certain conditional probabilities. It can be used to describe the impact of new data on the probability that a hypothesis is true. See David H. Kaye & Hal S. Stern, Reference Guide on Statistics and Research Methods, in this manual.
bin, fixed. In VNTR profiling, a bin is a range of base pairs (DNA fragment lengths). When a database is divided into fixed bins, the proportion of bands within each bin is determined and the relevant proportions are used in estimating the profile frequency.
bin, floating. In VNTR profiling, a bin is a range of base pairs (DNA fragment lengths). In a floating bin method of estimating a profile frequency, the bin is centered on the base-pair length of the allele in question, and the width of the bin can be defined by the laboratory’s matching rule (e.g., ±5% of band size).
binning. Grouping VNTR alleles into sets of similar sizes because the alleles’ lengths are too similar to differentiate.
biogeographic ancestry testing (BGA, BGAT). Inferring the geographic region where a person’s ancestors lived from DNA variants. Clinical and anthropological research studies often use thousands of SNPs for this purpose. Much smaller panels of these ancestry-informative markers have been developed for biological trace evidence.
blind proficiency test. See proficiency test.
capillary electrophoresis. A method for separating DNA fragments (including STRs) according to their lengths. A long, narrow tube is filled with an entangled polymer or comparable sieving medium, and an electric field is applied to pull DNA fragments placed at one end of the tube through the medium. The procedure is faster and uses smaller samples than gel electrophoresis, and it can be automated.
ceiling method. A procedure for setting a minimum DNA profile frequency proposed in 1992 by a committee of the National Academy of Sciences but not recommended by a second committee in 1996. One hundred persons from each of 15 to 20 genetically homogeneous populations spanning the range of racial groups in the United States are sampled. For each allele, the higher frequency among the groups sampled (or 5%, whichever is larger) is used in calculating the profile frequency. The committees called this procedure, intended to overstate the population frequency of a profile, the ceiling principle. Compare interim ceiling principle.
centimorgan (cM). A unit of measure for the frequency of genetic recombination. One centimorgan is equal to a 1% chance that two markers on a chromosome will become separated from one another owing to a recombination event during meiosis (which occurs during the formation of egg and sperm cells).
chip. A miniaturized system for genetic analysis. One such microfluidic chip mimics capillary electrophoresis and related manipulations. DNA fragments, pulled by small voltages, move through tiny channels etched into a small block of glass, silicon, quartz, or plastic. This system is useful in analyzing STRs. Another technique places a dense array of oligonucleotide probes on a solid surface. Such hybridization microarrays are useful in identifying SNPs and in sequencing mitochondrial DNA.
chromosome. A rodlike structure composed of DNA, RNA, and proteins. Most normal human cells contain 46 chromosomes, 22 autosomes and a sex chromosome (X) inherited from the mother, and another 22 autosomes and a sex chromosome (either X or Y) inherited from the father. The genes are located along the chromosomes. See also homologous chromosomes.
coding and noncoding DNA. The sequence of base pairs in a gene that correspond to the building blocks (amino acids) of a protein is called coding DNA. (A sequence of three base pairs, called a codon, specifies a particular one of the 20 possible amino acids in the protein. The mapping of a set of three nucleotide bases to a particular amino acid is the genetic code. The cell makes the protein through intermediate steps involving coding RNA transcripts.) About 1.5% of the human genome codes for the amino-acid
sequences. Another 23.5% of the genome is classified as genetic sequence but does not encode proteins. This portion of the noncoding DNA is involved in regulating the activity of genes. It includes promoters, enhancers, and repressors. Other gene-related DNA consists of introns (that interrupt the coding sequences, called exons, in genes and that are edited out of the RNA transcript for the protein), pseudogenes (evolutionary remnants of once-functional genes), and gene fragments. The remaining extragenic DNA (about 75% of the genome) also is noncoding.
CODIS (combined DNA index system). A collection of databases on STR and other loci of DNA from convicted felons, arrestees, missing persons, and crime scenes maintained by the FBI.
complementary sequence. The sequence of nucleotides on one strand of DNA that corresponds to the sequence on the other strand. For example, if one sequence is CTGAA, the complementary bases are GACTT.
control region. See D-loop.
cytoplasm. A jelly-like material (80% water) that fills the cell.
cytosine (C). One of the four bases, or nucleotides, that make up the DNA double helix. Cytosine binds only to guanine. See nucleotide.
databank. A collection of DNA samples; a sample repository.
database. A computer-searchable collection of DNA profiles.
degradation. The breaking down of DNA by chemical or physical means.
denature, denaturation. The process of splitting, as by heating, two complementary strands of the DNA double helix into single strands in preparation for hybridization with biological probes.
deoxyribonucleic acid (DNA). The molecule that contains genetic information. DNA is composed of nucleotide building blocks, each containing a base (A, C, G, or T), a phosphate, and a sugar. These nucleotides are linked together in a double helix—two strands of DNA molecules paired up at complementary bases (A with T, C with G). See adenine, cytosine, guanine, thymine.
diploid number. See haploid number.
D-loop. A portion of the mitochrondrial genome known as the control region or displacement loop that is instrumental in the regulation and initiation of mtDNA gene products. Two short “hypervariable” regions within the D-loop that do not appear to be functional are used in identity or kinship testing.
DNA polymerase. The enzyme that catalyzes the synthesis of double-stranded DNA.
DNA probe. See probe.
DNA profile. A list of the alleles at each locus. See genotype.
DNA sequence. The ordered list of base pairs in a duplex DNA molecule or of bases in a single strand.
DQ. The antigen that is the product of the DQA gene. See DQA, human leukocyte antigen.
DQA. The gene that codes for a particular class of human leukocyte antigen (HLA). This gene has been sequenced completely and can be used for forensic typing. See human leukocyte antigen.
electropherogram. The PCR products separated by capillary electrophoresis can be labeled with a dye that glows at a given wavelength in response to light shined on it. As the tagged fragments pass the light source, an electronic camera records the intensity of the fluorescence. Plotting the intensity as a function of time produces a series of peaks, with the shorter fragments producing peaks sooner. The intensity is measured in relative fluorescent units and is proportional to the number of glowing fragments passing by the detector. The graph of the intensity over time is an electropherogram.
electrophoresis. See capillary electrophoresis, gel electrophoresis.
endonuclease. An enzyme that cleaves the phosphodiester bond within a nucleotide chain.
environmental insult. Exposure of DNA to external agents such as heat, moisture, and ultraviolet radiation, or chemical or bacterial agents. Such exposure can interfere with the enzymes used in the testing process or otherwise make DNA difficult to analyze.
enzyme. A protein that catalyzes a reaction (speeds it up without being consumed).
epigenetic. Heritable changes in phenotype (appearance) or gene expression caused by mechanisms other than changes in the underlying DNA sequence. Epigenetic marks are molecules attached to DNA that can determine whether genes are active and used by the cell.
ethidium bromide. A molecule that can intercalate into DNA double helices when the helix is under torsional stress. Used to identify the presence of DNA in a sample by its fluorescence under ultraviolet light.
exon. See coding and noncoding DNA.
fallacy of the transposed conditional. See transposition fallacy.
false match. Two samples of DNA that have different profiles could be declared to match if, instead of measuring the distinct DNA in each sample, there is an error in handling or preparing samples such that the DNA from a single sample is analyzed twice. The resulting match, which does not reflect the true profiles of the DNA from each sample, is a false match. Some people use “false match”
more broadly, to include cases in which the true profiles of each sample are the same, but the samples come from different individuals. Compare true match. See also match, random match.
forensic DNA phenotyping (FDP). Inferring observable traits of individuals whose DNA is in a trace-evidence sample, or unknown deceased (missing) persons, directly from biological materials found at the scene. FDP is usually done with panels of SNPs in genes that influence external visible traits such as eye and hair color, but the term may include biogeographic ancestry testing.
forensic genetic genealogy (FGG). Another term for investigative genetic genealogy.
gel, agarose. A semisolid medium used to separate molecules by electrophoresis.
gel electrophoresis. In RFLP analysis, the process of sorting DNA fragments by size by applying an electric current to a gel. The different-sized fragments move at different rates through the gel.
gene. A set of nucleotide base pairs on a chromosome that contains the “instructions” for the product that controls some cellular function such as making an enzyme. The gene is the fundamental unit of heredity; each simple gene “codes” for a specific biological characteristic.
gene frequency. The relative frequency (proportion) of an allele in a population.
genetic drift. Random fluctuation in a population’s allele frequencies from generation to generation.
genetic genealogy. Determining family history (biological relationships between or among individuals) using DNA test results in combination with traditional genealogical methods. Direct-to-consumer genetic testing companies analyze DNA samples at hundreds of thousands of single-nucleotide polymorphisms spread across the genome. The locations and lengths of shared haploblocks are used to ascertain the degree of relatedness, and documentary and other research leads to the reconstruction of relevant parts of a family tree.
genetics. The study of the patterns, processes, and mechanisms of inheritance of biological characteristics.
genome. The complete genetic makeup of an organism, including roughly 23,000 genes and many other DNA sequences in humans. The haploid human genome comprises over three billion nucleotide base pairs.
genotype. The particular forms (alleles) of a set of genes possessed by an organism (as distinguished from phenotype, which refers to how the genotype expresses itself, as in physical appearance). In DNA analysis, the term is applied to the variations within one or all DNA regions (whether or not they constitute genes) that are analyzed.
genotype, multilocus. The alleles that an organism possesses at several sites in its genome.
genotype, single locus. The alleles that an organism possesses at a particular site in its genome.
guanine (G). One of the four bases, or nucleotides, that make up the DNA double helix. Guanine binds only to cytosine. See nucleotide.
haploblock (haplotype block). Several neighboring, tightly linked SNPs inherited together as a block. A haploblock has a higher discrimination power than any individual SNP within the block.
haploid number. Human sex cells (egg and sperm) contain 23 chromosomes each. This is the haploid number. When a sperm cell fertilizes an egg cell, the number of chromosomes doubles to 46. This is the diploid number.
haplotype. A specific combination of linked alleles at several loci on one chromosome. “Linked” means that the alleles tend to be inherited together because they are close to each other on the chromosome.
Hardy-Weinberg equilibrium. A condition in which the allele frequencies within a large, random, intrabreeding population are unrelated to patterns of mating. In this condition, the occurrence of alleles from each parent will be independent and have a joint frequency estimated by the product rule. See independence. Compare linkage disequilibrium.
heteroplasmy, heteroplasty. The condition in which some copies of mitochondrial DNA sequences in the same individual have different base pairs or lengths.
heterozygous. Having a different allele at a given locus on each of a pair of homologous chromosomes. See allele. Compare homozygous.
homologous chromosomes. The 44 autosomes (nonsex chromosomes) in the normal human genome are in homologous pairs (one from each parent) that share an identical set of genes, but may have different alleles at the same loci.
homozygous. Having the same allele at a given locus on each of a pair of homologous chromosomes. See allele. Compare heterozygous.
human leukocyte antigen (HLA). Antigen (foreign body that stimulates an immune system response) located on the surface of most cells (excluding red blood cells and sperm cells). HLAs differ among individuals and are associated closely with transplant rejection. See DQA.
hybridization. Pairing up of complementary strands of DNA from different sources at the matching base-pair sites. For example, a primer with the sequence AGGTCT would bond with the complementary sequence TCCAGA on a DNA fragment.
identical by descent (IBD). Haplotypes that are identical and inherited from a common ancestor. IBD blocks are broken up by recombination during meiosis, so their expected length depends on the number of generations since the common ancestor at the locus. If the common ancestor lived a great many generations ago (ancient IBD), the individuals share very short tracts of genetic material. At the other extreme, in families, individuals who have IBD typically share very long tracts (greater than 10 centimorgans), and IBD is only defined with respect to documented common ancestry. Recent IBD is IBD between individuals of possibly undocumented relationship, and it results from common ancestry within approximately the past 30 generations.
INDEL. A general term that may refer to insertion or deletion of nucleotides in genomic DNA. Compare single nucleotide polymorphism.
independence. Two events are said to be independent if one is neither more nor less likely to occur when the other does.
investigative genetic genealogy (IGG). The application of genetic genealogy to generate a list of individuals whose DNA might match that of a trace-evidence or missing-person DNA sample. Also called forensic genetic genealogy (FGG) or forensic investigative genetic genealogy (FIGG).
interim ceiling principle. A procedure proposed in 1992 (and no longer used) by a committee of the National Academy of Sciences for setting a minimum DNA profile frequency. For each allele, the highest frequency (adjusted upward for sampling error) found in any major racial group (or 10%, whichever is higher) is used in product-rule calculations. Compare ceiling principle.
intron. See coding and noncoding DNA.
kilobase (kb). A measure of DNA length (1,000 bases).
kinship analysis. Evaluation of biological relatedness between individuals based on DNA. Kinship analysis is used in parentage testing (civil or criminal), disaster victim identification, missing persons identification, familial searching, genetic genealogy, and immigration control.
kinship index. A likelihood ratio used for kinship analysis. See paternity index.
likelihood ratio (LR). A measure of the support that an observation provides for one hypothesis as opposed to an alternative hypothesis. The likelihood ratio is computed by dividing the conditional probability of the observation given that one hypothesis is true by the conditional probability of the observation given the alternative hypothesis. For example, the likelihood ratio for the hypothesis that two DNA samples with the same correctly ascertained STR profile originated from the same individual (as opposed to originating from two unrelated individuals) is the reciprocal of the
random-match probability. Legal scholars have introduced the likelihood ratio as a measure of the probative value of evidence. Evidence that is 100 times more probable to be observed when one hypothesis is true as opposed to another has more probative value than evidence that is only twice as probable.
lineage marker. DNA features that are inherited exclusively from one parent. Lineage markers are found on sex chromosomes and in mitochondrial DNA.
linkage. The inheritance together of two or more genes or loci on the same chromosome.
linkage equilibrium. A condition in which the occurrence of alleles at different loci is independent.
locus. A location in the genome, that is, a position on a chromosome where a gene, marker, or other structure begins.
Markov chain Monte Carlo approximation. A computer simulation method that can be used to get an approximate solution for the integration that must be performed to apply Bayes’ rule to a problem with many random variables. It is used in some computer programs that give ratios of probabilities of the data given the different sets of genotypes that might be present in a DNA sample.
massively parallel sequencing (MPS). Any of several high-throughput methods for determining nearly all or just parts of the nucleotide sequence of an individual’s genome. Multiple DNA segments are analyzed simultaneously. There are several instruments with different designs to accomplish this. Also called next-generation sequencing (NGS). See reads.
match. The presence of the same allele or alleles in two samples. Two DNA profiles are declared to match when they are indistinguishable in genotype. For loci such as STRs, with discrete alleles, two samples match when they display the same set of alleles. For RFLP testing of VNTRs, two samples match when the pattern of the bands is similar and the positions of the corresponding bands at each locus fall within a preset distance. See match window, false match, true match.
match window. If two RFLP bands lie within a preset distance (called the match window) that reflects normal measurement error, the bands can be declared to match.
meiosis. A special type of cell division of germ cells that produces sperm or egg cells with only one copy of each chromosome (a haploid genome). In humans, body (somatic) cells are diploid, containing two sets of these chromosomes (one from each parent via the mother’s egg cell and the father’s sperm cell). Compare mitosis.
messenger RNA (mRNA). A type of single-stranded RNA that carries information from the DNA in a cell’s nucleus to the cell’s watery interior, where the protein-making machinery reads the mRNA sequence and translates it into the amino acid in a growing protein chain. See coding and noncoding DNA.
methylation. A chemical reaction in the body in which a small molecule called a methyl group gets added to DNA, proteins, or other molecules. The addition of methyl groups can affect how some molecules act in the body. Methylation turns gene expression on or off. See epigenetic.
microarray. See chip.
microfluidics. The technology of manufacturing microminiaturized devices containing chambers and tunnels through which fluids flow or are confined.
microhaplotype. Microhaplotype loci (microhaps) are markers less than 300 nucleotides long that display multiple allelic combinations (polymorphism).
microsatellite. Another term for an STR.
minisatellite. Another term for a VNTR.
mitochondria. A structure (organelle) within nucleated (eukaryotic) cells that is the site of the energy-producing reactions within the cell. Mitochondria contain their own DNA (often abbreviated as mtDNA), which is inherited only from mother to child.
mitosis. The process by which a cell replicates its chromosomes and then segregates them, producing two identical nuclei in preparation for cell division. Mitosis is generally followed by equal division of the cell’s content into two daughter cells that have identical genomes. The copying of DNA from a human somatic (body) cell that splits to produce a daughter cell occurs in mitosis. Compare meiosis.
molecular weight. The weight in grams of 1 mole (approximately 6.02 × 1023 molecules) of a pure, molecular substance.
monomorphic. A gene or DNA characteristic that is almost always found in only one form in a population. Compare polymorphism.
multilocus probe. A probe that marks multiple sites (loci). RFLP analysis using a multilocus probe will yield an autorad showing a striped pattern of thirty or more bands. Such probes are no longer used in forensic applications.
multilocus profile. See profile.
multiplexing. Typing several loci simultaneously.
mutation. The process that produces a gene or chromosome set differing from the type already in the population; the gene or chromosome set that results from such a process.
nanogram (ng). A billionth of a gram.
next-generation sequencing (NGS). Any implementation of massively parallel sequencing. There are several generations of NGS. See reads.
nucleic acid. RNA or DNA.
nucleotide. A unit of DNA consisting of a base (A, C, G, or T) and attached to a phosphate and a sugar group; the basic building block of nucleic acids. See deoxyribonucleic acid.
nucleus. The membrane-covered portion of a eukaryotic cell containing most of the DNA and found within the cell’s watery interior (the cytoplasm).
oligonucleotide. A synthetic polymer made up of fewer than 100 nucleotides; used as a primer or a probe in PCR. See primer.
paternity index. A number (technically, a likelihood ratio) that indicates the support that the paternity test results lend to the hypothesis that the alleged father is the biological father as opposed to the hypothesis that another man selected at random is the biological father. Assuming that the observed DNA genotypes (or phenotypes) correctly represent those of the mother, child, and alleged father tested, the number can be computed as the ratio of the probability of the genotypes (or phenotypes) under the first hypothesis to the probability under the second hypothesis. The paternity index is one type of kinship index; the siblingship index is another.
pH. A measure of the acidity of a solution.
phenotype. An observable trait, such as height, eye color, or blood group, resulting from a genotype and environmental factors.
picogram. A trillionth of a gram. A human cell contains about six picograms of DNA.
point mutation. See SNP.
polymarker. A commercially marketed set of PCR-based DNA tests for certain protein polymorphisms.
polymerase chain reaction (PCR). A process that mimics DNA’s own replication processes to make up to millions of copies of short strands of genetic material in a few hours.
polymorphism. The presence of several forms of a gene or DNA characteristic in a population. Mutations give rise to polymorphisms. In a genome sequencing project, single nucleotide polymorphisms (SNPs) and DNA mutations are defined as DNA variants detectable in more than 1% or in less than 1% of the population, respectively. Compare allele.
population genetics. The study of the genetic composition of groups of individuals.
population structure. When a population is divided into subgroups that do not mix freely, that population is said to have structure. Significant structure can lead to allele frequencies being different in the subpopulations.
primer. An oligonucleotide that attaches to one end of a DNA fragment and provides a point for more complementary nucleotides to attach and replicate the DNA strand. See oligonucleotide.
probabilistic genotyping. Electropherograms display patterns of peaks that could result from different genotypes. Each possible genotype has its own probability of giving rise to the observed pattern (the data). In traditional binary matching, every genotype is said to match with a probability of 0 or 1, and a likelihood ratio for the single matching genotype (or single set of genotypes in a mixed profile) is assigned. Probabilistic genotyping allows the possible genotypes to give rise to the data with probabilities between 0 and 1. It uses statistical modeling to compute these probabilities. Likelihood ratios are reported for the possible genotypes (or combinations of genotypes that could be in a mixed profile).
probe. In forensics, a short segment of DNA used to detect certain alleles. The probe hybridizes, or matches up, to a specific complementary sequence. Probes allow visualization of the hybridized DNA, either by a radioactive tag (usually used for RFLP analyses) or a biochemical tag (usually used for PCR-based analyses).
product rule. When alleles occur independently at each locus (Hardy-Weinberg equilibrium) and across loci (linkage equilibrium), the proportion of the population with a given genotype is the product of the proportion of each allele at each locus, times factors of two for heterozygous loci.
proficiency test. A test administered at a laboratory to evaluate its performance. In a blind proficiency study, the laboratory personnel do not know that they are being tested.
prosecutor’s fallacy. See transposition fallacy.
protein. Biologically important molecules made up of a linear string of building blocks called amino acids. The order in which these components are arranged is encoded in the DNA sequence of the gene that expresses the protein. See coding DNA.
pseudogenes. Genes that have been so disabled by mutations that they can no longer produce proteins. Some pseudogenes can still produce noncoding RNA.
quality assurance. A program conducted by a laboratory to ensure accuracy and reliability.
quality audit. A systematic and independent examination and evaluation of a laboratory’s operations.
quality control. Activities used to monitor the ability of DNA typing to meet specified criteria.
random match. A match between a specific DNA profile and a second DNA profile drawn at random from the population. See also random-match probability.
random-match probability. The chance of a random match. As it is usually used in court, the random-match probability refers to the probability of a true match when the DNA being compared to the evidence DNA comes from a person drawn at random from the population. This random-true-match probability reveals the probability of a true match when the samples of DNA come from different, unrelated people.
random mating. The members of a population are said to mate randomly with respect to particular genes of DNA characteristics when the choice of mates is independent of the alleles.
rapid DNA. A fully automated process of developing a DNA profile from a sample without the need for a DNA laboratory or human intervention. A microfluidic lab-on-a-chip and associated software perform the extraction, amplification, separation, detection, and allele calling.
reads. Using next-generation “short-read” sequencing, DNA is broken into short fragments (typically 75–300 base pairs) that are amplified (copied) and then sequenced to produce “reads.” Bioinformatic techniques then arrange the reads into a continuous genomic sequence. “Third-generation sequencing” methods permit “long-read” sequencing (> 10,000 bp).
recombination. In general, any process in a diploid or partially diploid cell that generates new gene or chromosomal combinations not found in that cell or in its progenitors.
reference population. The population to which the source of a trace-evidence sample is thought to belong.
relative fluorescent unit (RFU). See electropherogram.
replication. The synthesis of new DNA from existing DNA. See polymerase chain reaction.
restriction enzyme. Protein that cuts double-stranded DNA at specific base-pair sequences (different enzymes recognize different sequences). See restriction site.
restriction fragment length polymorphism (RFLP). Variation among people in the length of a segment of DNA cut at two restriction sites.
restriction fragment length polymorphism (RFLP) analysis. Analysis of individual variations in the lengths of DNA fragments produced by digesting sample DNA with a restriction enzyme.
restriction site. A sequence marking the location at which a restriction enzyme cuts DNA into fragments. See restriction enzyme.
reverse dot blot. A detection method used to identify SNPs in which DNA probes are affixed to a membrane, and amplified DNA is passed over the probes to see if it contains the complementary sequence.
ribonucleic acid (RNA). A single-stranded molecule “transcribed” from DNA. “Coding” RNA acts as a template for building proteins according to the sequences in the coding DNA from which it is transcribed. Other RNA transcripts can be a sensor for detecting signals that affect gene expression or can be a switch for turning genes off or on, or they may be functionless.
sequence-specific oligonucleotide (SSO) probe. Also, allele-specific oligonucleotide (ASO) probe. Oligonucleotide probes used in a PCR-associated detection technique to identify the presence or absence of certain base-pair sequences identifying different alleles. The probes are visualized by an array of dots rather than by the electropherograms associated with STR analysis.
sequencing. Determining the order of base pairs in a segment of DNA or RNA.
short tandem repeat (STR). A class of repetitive DNA resulting from multiple copies of virtually identical base-pair sequences, arranged in succession at a specific locus on a chromosome. The number of repeats varies from individual to individual. STRs are shorter than VNTRs.
single-locus probe. A probe that only marks a specific site (locus). A single-locus probe for a VNTR will produce one or two bands in an autoradiograph. Compare multilocus probe.
SNP (single nucleotide polymorphism). A substitution, insertion, or deletion of a single base pair at a given point in the genome. See polymorphism. Compare indel.
SNP chip. See chip.
Southern blotting. Named for its inventor, a technique by which processed DNA fragments, separated by gel electrophoresis, are transferred onto a nylon membrane in preparation for the application of biological probes.
thymine (T). One of the four bases, or nucleotides, that make up the DNA double helix. Thymine binds only to adenine. See nucleotide.
transcription. The process by which the information in a strand of DNA is copied into a new molecule of messenger RNA (mRNA). The mRNA transcript serves as a blueprint for protein synthesis during the process of translation. See messenger RNA.
transposition fallacy. Also called the prosecutor’s fallacy, the transposition fallacy confuses the conditional probability of A given B with that of B given A.
Few people think that the probability that a person speaks Spanish given that he or she is a citizen of Chile equals the probability that a person is a citizen of Chile given that he or she speaks Spanish. Yet many court opinions, newspaper articles, and even some expert witnesses speak of the probability of a matching DNA genotype given that someone other than the defendant is the source of the crime-scene DNA as if it were the probability of someone else being the source given the matching profile. Transposing conditional probabilities correctly requires Bayes’ rule.
true match. Two samples of DNA that have the same profile should match when tested. If there is no error in the labeling, handling, and analysis of the samples and in the reporting of the results, a match is a true match. A true match establishes that the two samples of DNA have the same profile. Unless the profile is unique, however, a true match does not conclusively prove that the two samples came from the same source. Some people use “true match” more narrowly, to mean only those matches among samples from the same source. Compare false match. See also match, random match.
variable number tandem repeat (VNTR). A class of RFLPs resulting from multiple copies of virtually identical base-pair sequences, arranged in succession at a specific locus on a chromosome. The number of repeats varies from individual to individual, thus providing a basis for individual recognition. VNTRs are longer than STRs.
whole genome sequencing. Determining the order of all the nucleotides in an individual organism’s DNA. Usually accomplished with a massively parallel sequencing system.
window. See match window.
X chromosome. See chromosome.
Y chromosome. See chromosome.
References on DNA
Genetics and Genomics
John M. Archibald, Genomics: A Very Short Introduction (2018).
Jocelyn E. Krebs et al., Lewin’s Genes XII (2018).
James D. Watson et al., Molecular Biology of the Gene (7th ed. 2014).
Forensic Genetics and Genomics
Jo-Anne Bright & Michael D. Coble, Forensic DNA Profiling: A Practical Guide to Assigning Likelihood Ratios (2020).
Forensic DNA Interpretation (John Buckleton et al. eds., 2d ed. 2016).
John M. Butler, Fundamentals of Forensic DNA Typing (2009); Advanced Topics in Forensic DNA Typing: Methodology (2011); Advanced Topics in Forensic DNA Typing: Interpretation (2015).
Silent Witness: Forensic DNA Analysis in Criminal Investigations and Humanitarian Disasters (Henry Ehrlich et al. eds., 2020).
Ian W. Evett & Bruce S. Weir, Interpreting DNA Evidence: Statistical Genetics for Forensic Scientists (1998).
William Goodwin et al., An Introduction to Forensic Genetics (2d ed. 2011).
David H. Kaye, The Double Helix and the Law of Evidence (2010).
Erin E. Murphy, Inside the Cell: The Dark Side of Forensic DNA (2015).
National Research Council Committee on DNA Forensic Science: An Update, The Evaluation of Forensic DNA Evidence (1996).
National Research Council Committee on DNA Technology in Forensic Science, DNA Technology in Forensic Science (1992).
Royal Soc’y, Forensic DNA Analysis: A Primer for Courts (2017).

























































































































































